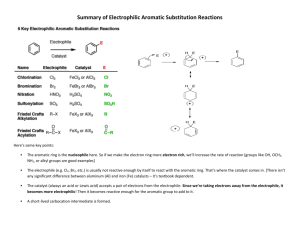
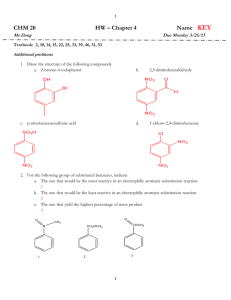
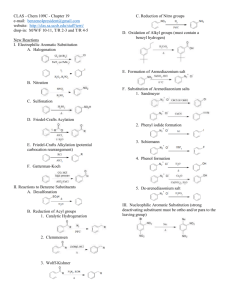
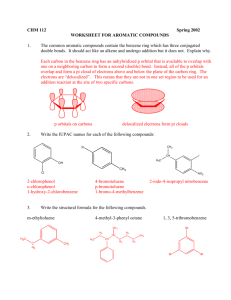
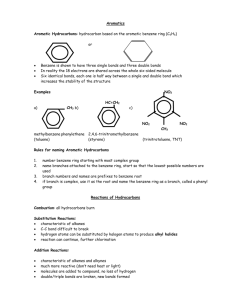
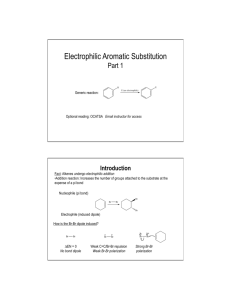
Topic 30: Electrophilic Aromatic Substitution
advertisement

Topic 30: Electrophilic Aromatic Substitution Note to students: This is a single chapter (or a portion of one) from a textbook that is under construction. Therefore you can ignore references to other textbook sections. 30.1 Why Should I Study This? In this topic we will explore a series of related reactions in which a hydrogen atom of an aromatic ring is substituted (replaced) by another atom or group. The mechanistic entity that brings about this substitution process is an electrophile (abbreviated as Elec), so we refer to this reaction as electrophilic aromatic substitution (EAS). Elec H Elec EAS is useful in to make new aromatic molecules, and therefore is commonly used in industrial-scale synthesis. For example, EAS is a key step in the conversion of toluene into 2,4,6-trinitrotoulene (better known as TNT, an explosive) and naphthalene into FD&C Red No. 40 (a red dye). Toluene and naphthalene are both available in huge quantities from petroleum. CH3 CH3 O2N NO2 EAS NO2 Toluene 2,4,6-Trinitrotoluene (TNT) _________________________________________________________ Electrophilic Aromatic Substitution 1 Electrophilic aromatic substitution: A reaction in which an electrophile adds to an aromatic ring, ultimately replacing an aromatic ring hydrogen atom. Na O3S OH N EAS N FD&C Red No. 40 OCH3 Naphthalene CH3 SO3 Na We will explore these reactions, as well as many others, in more detail as this topic unfolds. Although there are few (if any) examples of biochemical EAS reactions, this topic is worthy of our time and effort because through its study we will uncover some interesting lessons about the influence of molecular structure on a reaction’s mechanism, rate and products. 30.2 Why is Benzene Different? Substitution versus Addition Note that the electrophile is just a protonated leaving group, and that the leaving group functions as a base in a later mechanism step. We saw this pattern other mechanisms: SN1 (section 25.xx), E1 (section 27.xx) and some pi bond addition reactions (sections 28.xx), and we will see it again. Let’s start with a very basic but very important observation about EAS reactions. A benzene ring has pi electrons, so in analogy to what we learned in Topic 28, we expect it to react with electrophiles. The reaction of an alkene such as cyclohexene (a nucleophile) with strong aqueous acid (H3O+, an electrophile) gives cyclohexanol, an addition product. H2O H OH2 OH2 H OH OH Benzene has six pi electrons, so by analogy, we expect benzene to behave much like cyclohexene. Think Ahead Question 30.1 By working through the reaction mechanism, predict the product formed by the reaction of benzene with H3O+. Answer: The predicted mechanism looks very much like the cyclohexene hydration reaction above, but the nucleophile has two more double bonds. _________________________________________________________ Electrophilic Aromatic Substitution 2 H2O H OH2 H OH OH2 OH Arenium ion The carbocation derived from electrophilic attack on the benzene ring is called an arenium ion. (Arene is an old term for aromatic molecules like benzene.) An arenium ion is also called a σ complex or a Wheland intermediate. Based on this mechanism, we predict the reaction product is cyclohexa-2,4-dien-1-ol. However, this is just a prediction, and as we have seen many times before, Mother Nature sometimes has her own ideas. When we run the reaction in the lab in order to test the prediction, we discover the reaction gives substitution products, and not addition products. For example, heating benzene with a mixture of aqueous sulfuric and nitric acids gives nitrobenzene (a substitution product) and not the alcohol product expected by an addition mechanism. OH aq. HNO3/H2SO4 NO2 or Addition product expected but not observed Substitution product actual product Lets explore the reason why benzene undergoes substitution instead of addition by examining the mechanism of a very similar reaction. The reaction involves introduction of deuterium (2H or D), a hydrogen isotope that behaves almost exactly like the more familiar hydrogen isotope 1H in a reaction mechanism, yet it can be differentiated from 1 H in a molecule.* For example, D3O+ is the deuterium analog of H3O+. Deuterium can help us track proton transfers during a reaction, and draw conclusions about the reaction mechanism. Think Ahead Question 30.2 Select the major product when an excess of benzene is reacted with D3O+, formed in a mixture of D2SO4 and D2O. * There is one important difference in the mechanism behavior of hydrogen and deuterium. Bonds to deuterium are stronger than bonds to hydrogen, so breaking a bond to deuterium has a larger ΔG‡. This influences the reaction rate, and is called a kinetic isotope effect. _________________________________________________________ Electrophilic Aromatic Substitution 3 Arenium ion: A carbocation formed by electrophilic attack on a benzene ring. D OD D2O/D2SO4 or Answer: In the first mechanism step, D3O+ (the electrophile) attacks the benzene ring to give an arenium ion. D H OD2 H D H D D An arenium ion is just another type of carbocation, so now we consider carbocation fates. The carbocation can capture a nucleophile, be deprotonated or rearrange. Recall that carbocations are generally highly unstable and very reactive, and so they are not very selective with which carbocation pathway they use to achieve stability. (Review section 24.xx if you don’t feel confident with carbocation fates.) However, in this case deprotonation restores aromaticity whereas capture of water does not. Deprotonation has an extra thermodynamic incentive over the other carbocation fates. (Recall from section 29.xx that for benzene, aromaticity is worth 36 kcal mol-1 of extra stability.) Therefore deuterobenzene is the major product. Capture nucleophile: D2O D D OD O D2O H H H D D D + D2O D Aromaticity not restored - Disfavored pathway Deprotonation: H OD2 D D + H OD2 Aromaticity restored - Favored pathway (Deprotonation of the deuterium instead of the hydrogen also restores aromaticity, so it is also favored. However, because this pathway regenerates benzene we cannot detect its occurrence.) _________________________________________________________ Electrophilic Aromatic Substitution 4 Because aromatic molecules have significantly less free energy than similar molecules that are not aromatic, there is a strong driving force to gain aromaticity whenever possible. For example, cyclohexa-2,4dien-1-ol quickly loses water to form benzene in order to gain aromaticity. OH + H2O 30.3 A General Mechanism for Electrophilic Aromatic Substitution Electrophilic aromatic substitution reactions all achieve the same change in molecular structure: some other atom or group replaces hydrogen atom on an aromatic ring. So you might reasonably assume that the mechanisms for all these reactions also have quite a bit in common. (We have encountered this general principle many times before. Similar functional group transformations often have similar reaction mechanisms.) Think Ahead Question 30.3 write a general mechanism for a generic EAS reaction of an electrophile (Elec) with benzene (first reaction in this topic), based on the reactions in section 30.2. Answer: As we have seen in section 30.2, the EAS mechanism is very much like the addition of certain electrophiles to alkenes or alkynes. The first step is electrophilic attack on the pi electrons, to form a carbocation. Now we ask ourselves the usual carbocation question: Which carbocation fate happens? As discussed in section 30.2, deprotonation of the arenium ion has a great thermodynamic advantage over nucleophile capture or rearrangement because deprotonation restores aromaticity (worth an extra 36 kcal mol-1 in benzene!) whereas the other carbocation fates do not. _________________________________________________________ Electrophilic Aromatic Substitution 5 General Mechanism: Electrophilic Aromatic Substitution Resonance contributors H B Elec Elec Elec Electrophile attacks benzene ring forming resonance - stabilized arenium ion. Carbocation fate: deprotonation. Only this carbocation fate restores aromaticity. The formal charges on the electrophile and base are not specified. This is because our mechanism is a generalization, and we do not know the exact structure of the electrophile or base. Without knowing their structures we cannot assign formal charges. (Like all electrophiles, Elec carries a δ+ or formal +1 charge. Similarly, B carries a δ- or formal –1 charge.) Ipso hydrogen: In an electrophilic aromatic substitution, the hydrogen attached to the carbon that is attacked by the incoming electrophile. Avoid This Common Problem Hydrogen atoms are often not shown in skeletal structures (section 1.xx), but the hydrogen on the sp3 carbon atom that bears the electrophile (called the ipso hydrogen) is shown explicitly. Why does this hydrogen get special treatment? Sometimes students forget about this hydrogen and think the carbon has an open octet. This leads to all sorts of mechanism mistakes. Always drawing the ipso hydrogen will help you avoid this common pitfall. Ipso hydrogen H Elec The arenium ion has resonance. As we shall see this resonance plays a key role when an EAS reaction can give more than one product. Think Ahead Question 30.4 Draw all of the important resonance contributors, plus the resonance hybrid for the arenium ion in our general EAS mechanism. How does this resonance influence the choice of major product and the reaction rate? (Resonance is an important issue in EAS, so if you don’t feel confident in your ability to draw these resonance contributors, review section 5.4.) _________________________________________________________ Electrophilic Aromatic Substitution 6 Answer: The three important resonance contributors plus the hybrid are shown. H H Elec H Elec Resonance contributors H Elec Elec Resonance hybrid How does resonance influence the choice of major product? Recall the useful assumption that the more stable product or reaction intermediate is the major one (thermodynamic control; section 20.xx). So if more than one product is possible, the major product will be derived from the more stable arenium ion (unless the reaction is under kinetic control). How does resonance influence the rate of the reaction? Arenium ion resonance delocalizes the formal positive charge. This stabilizes the structure and lowers the energy of activation required to form it. Lower energy of activation results in a faster rate for that step of the mechanism. But we cannot also say that it will result in a faster rate for the entire reaction, because the general EAS mechanism has two steps (or more if additional steps are required to form the electrophile) and we do not know which is the rate-determining step (rds). As we have seen numerous times before, it can be useful to explore the features that influence a reaction’s rate, and EAS is no exception. Therefore we need to determine which mechanism step is the rds. Think Ahead Question 30.5 Which step of the general EAS mechanism is the rate-determining step? Draw an energy profile for the general EAS reaction. Assume the reaction is slightly exergonic. Answer: The general mechanism includes two steps (electrophilic attack to form the arenium ion and subsequent deprotonation of the arenium ion), each with its own transition state and rate, so the energy profile has two hills and two free energies of activation (ΔG‡). The rate-determining (slowest) mechanism step has the largest ΔG ‡. (Review section 20.xx if you are a bit fuzzy about any of these concepts.) In most EAS reactions, several mechanism steps are necessary to produce the electrophile that actually attacks the benzene ring. For this discussion we will assume these steps are not rate determining. _________________________________________________________ Electrophilic Aromatic Substitution 7 (Kinetic studies suggest this assumption is probably true in many, if not all EAS reactions.) When we consider the thermodynamics of the two mechanism steps, one feature is obvious. Electrophilic attack on the benzene ring (first step) disrupts aromaticity. Deprotonation of the arenium ion (second step) restores aromaticity. Aromaticity is a significant stabilizing feature, and its loss is energetically expensive. Thus we expect ΔG‡ for the electrophilic attack step to be higher than Δ G ‡ for the deprotonation step. By this logic we predict that electrophilic attack is the rate-determining step. (This analysis ignores other energy changes such as loss and formation of bonds. Experimental kinetic evidence confirms that the electrophilic attack step is rate determining. This suggests that either aromaticity is in fact the deciding thermodynamic factor or the bond energy analysis gives the same result.) In General... Compare the rate-determining step for EAS, SN1 and E1. Do you see a pattern? In each case, the mechanism step in which the carbocation is formed is rate determining. This pattern applies to many other reactions as well. So as a general assumption we can say that carbocation formation is often the rate-determining mechanism step. Now that we know the rate-determining step we can draw the energy profile. H Elec δ+ H B Elec Energy δ+ H ΔG1 rds Elec ΔG2 Elec Reaction coordinate Now that we have developed a general EAS mechanism and have an idea of its kinetics, we can explore specific cases. Because EAS is _________________________________________________________ Electrophilic Aromatic Substitution 8 such a valuable reaction for introducing new substituents to a benzene ring, many different EAS reactions have been developed. We will examine a few of the more important reactions. We will also see patterns of reactivity, regioselectivity and mechanism that are common to all of these EAS reactions. Studying these reactions from the perspective of these patterns will ease your journey through this topic. 30.4 Halogenation A. Bromination EAS is an effective method to replace a benzene ring hydrogen with a halogen such as bromine. Br EAS Using what we know about electrophiles and EAS, we can deduce the how this reaction might be achieved. Think Ahead Question 30.6 What electrophile is needed to convert benzene to bromobenzene by the EAS mechanism? How might this electrophile be generated? Write an EAS mechanism for the conversion of benzene into bromobenzene. Answer: This EAS involves replacement of a hydrogen atom by a bromine atom, so by analogy to the general EAS mechanism (section 30.3) the electrophile is an electron-deficient bromine atom. This could be bromine bearing a leaving group (Br-LG). In fact, we have already seen Br2 react this way, when it adds to an alkene or alkyne (section 29.xx). So the reaction of Br2 with benzene might proceed this way: H Br Br Br Br Br So with great anticipation, we go into the lab and mix Br2 with benzene, expecting an instantaneous reaction (just like an Br2 with an alkene or alkyne). But nothing happens! _________________________________________________________ Electrophilic Aromatic Substitution 9 Why is Br2 less reactive with benzene than with an alkene? The answer is aromaticity. As we have seen before (section 30.3), addition of an electrophile to benzene is energetically expensive because aromaticity is lost. Bromine is sufficiently electrophilic to add to an alkene or alkyne (where aromaticity is not lost) but is just not powerful enough to overcome the higher activation energy needed to disrupt aromaticity. How do we make Br2 more electrophilic? One way is to make the leaving group depart more easily. Another way is to magnify the δ+ charge on one of the bromine atoms. (In other words, we want Br2 to have a permanent bond dipole instead of an induced bond dipole.) Both of these goals can be achieved by adding a strong Lewis acid such FeBr3. Being electron-deficient, the Lewis acid borrows a bromine lone pair to form a new bond, causing the bromine to have a formal positive charge. Bromine with a formal positive charge is more electronegative than neutral bromine, so a Br-Br bond dipole results. (You can also think of this as an inductive effect.) The Br-Br bond is also weakened. δ+ Br Br FeBr3 Lewis acid; accepts electrons Br Br FeBr3 δ+ due to Br+ With a larger δ+ on Br and a weakened Br-Br bond, the Br2-FeBr3 complex is sufficiently electrophilic to overcome the high activation energy caused by the benzene ring’s loss of aromaticity. FeBr3 is deliquescent (it absorbs enough water from the air to form a solution). Moist FeBr3 is not effective as an EAS catalyst. However, FeBr3 is easily generated in situ by reacting Fe and Br2, so we can write the overall reaction this way: Br Fe, Br2 Think Ahead Question 30.7 Write the EAS mechanism for the reaction of the Br2-FeBr3 complex with benzene. Answer: The reaction follows the general EAS mechanism we worked out before. _________________________________________________________ Electrophilic Aromatic Substitution 10 H Br FeBr3 Br Br Br δ+ + HBr + FeBr3 BrFeBr3 The FeBr4- functions as a base in order to neutralize the formal negative charge on iron. Recall that carbocations are unstable and highly reactive, so they can be deprotonated even by weak bases (section 20.xx). Compared to other carbocations, arenium ion deprotonation is exceptionally easy because aromaticity is restored. Note that FeBr3 is regenerated in the last mechanism step. It can react with another Br2 molecule, so it can be considered as a catalyst in this reaction. We can often draw two or more similar mechanisms for a reaction that vary somewhat in the sequence of bond breaking and bond forming. Your answer to Think Ahead Question 30.7 might be a bit different that the mechanism just presented. For example, the electrophile that attacked the benzene ring could be Br+ instead of the Br2-FeBr3 complex. Br Br + FeBr4 BrFeBr3 H Br Br Or you may have used Br- instead of FeBr4- to deprotonate the arenium ion. Br FeBr3 Br + FeBr3 H Br Br Br Both of these are reasonable mechanism alternatives. (The actual mechanism is dependent on the reactants that are used as well as other reaction variables such as solvent, temperature, etc. On paper we can accept any mechanism that is reasonable and consistent with the facts at hand.) _________________________________________________________ Electrophilic Aromatic Substitution 11 Concept Focus Question 30.1 Write the products and mechanism for this reaction. H3 C CH3 Br2, Fe Concept Focus Question 30.2 (a) Why is FeBr3 a Lewis acid? (b) Which one of these substances is not a Lewis acid: FeCl3, AlCl3, BF3, CCl4. B. Chlorination Now that we know how to introduce bromine onto a benzene ring by EAS, let’s consider the same transformation with other halogens. Think Ahead Question 30.8 What reagents might be used to convert benzene into chlorobenzene? Write a mechanism. Answer: We used Br2 and Fe to convert benzene into bromobenzene, so by analogy, Cl2 and Fe might useful to convert benzene into bromobenzene. The mechanism is also similar. Cl Fe, Cl2 Mechanism: EAS Chlorination of Benzene FeCl3 Fe + Cl2 δ+ Cl Cl FeCl3 Cl Cl FeCl3 H Cl FeCl3 Cl Cl Cl δ+ ClFeCl3 + HCl + FeCl3 _________________________________________________________ Electrophilic Aromatic Substitution 12 In parallel to the bromination mechanism, the electrophile could be Cl+ instead of the Cl2-FeCl3 complex, and the base could be Cl- instead of FeCl4-. C. Iodination and Fluorination Iodination of a benzene ring can also be accomplished by EAS. The electrophile is I+, which is conveniently generated from I2 by oxidation with HgO or HNO3. (An I2-Lewis acid complex is not sufficiently electrophilic.) I I2, HgO We might expect a benzene ring to be fluorinated by F+, but the high electronegativity of fluorine makes this impractical. Reagents for fluorination in which a fluorine atom bears a leaving group (F-LG) have been developed. However, the most effect strategy to introduce fluorine onto a benzene ring is to replace an existing benzene ring substituent (such as NH2) with fluorine (section 40.xx). Concept Focus Question 30.3 Give the product of this reaction: CH3 I2, HgO H3C Concept Focus Question 30.4 There is some evidence that conversion of benzene to iodobenzene with I2 and SO3 may involve I3+ instead of I+. Write a mechanism for the I3+ version of this EAS reaction. I I2, SO3 _________________________________________________________ Electrophilic Aromatic Substitution 13 Fluorine is considered the most reactive element, exothermically attacking almost everything it encounters, including glass. Only certain metal alloys or substances that are already highly fluorinated (such as Teflon) resist its aggressive nature. This aggressiveness makes F2 impractical for use in normal organic synthesis. 30.5 Substituent Effects: Electron-Donating Groups A. Directing Effects So far we have explored EAS reactions in cases where only a single reaction product is possible. In the vast majority of cases, however, more than one product might be formed. Think Ahead Question 30.9 EAS bromination of toluene with Br2 and Fe leads to more than one bromotoluene isomer. How many isomers are possible and what are their structures? (It may be useful at this point to review the nomenclature of substituted benzenes in section 6.xx.) Answer: There are three bromotoluene products, namely the ortho, meta and para isomers. CH3 CH3 CH3 CH3 Br Br2, Fe + + Br ortho-bromotoluene meta-bromotoluene Br para-bromotoluene (You might think that because there are five benzene ring hydrogens that there are also five possible products. Compare models of all five possibilities to determine which are in fact identical.) Which isomer is the major one? Several factors contribute to this. Think Ahead Question 30.10 Toluene has two hydrogen atoms that are ortho to the methyl group, two that are meta and just one that is para. How does this fact influence the ortho/meta/para product ratio? Answer: EAS involves replacement of a benzene ring hydrogen with some other atom or group. Ignoring all other factors, there is a greater chance that an ortho or meta hydrogen is replaced because there are two of each of these, but only one para hydrogen. Based just on this statistical analysis, the product will consist of 40% of the ortho isomer, 40% of the meat isomer and 20% of the para isomer. _________________________________________________________ Electrophilic Aromatic Substitution 14 CH3 Two ortho hydrogens H H Two meta hydrogens H H H One para hydrogen Think Ahead Question 30.11 How does steric hindrance influence the product ratio during the EAS bromination of toluene? It may be useful to examine a model of toluene. Answer: First, let’s review the basics of steric hindrance. For a new bond to form, the atoms in question must come within bond distance. This can be slowed or even prevented if the reactants have group(s) of atoms that block this close approach. For example, SN2 reactions do not occur at tertiary carbons because the alkyl groups prevent approach of the nucleophile. (Read section 22.xx if necessary to review the concept of steric hindrance.) Now we can rephrase Think Ahead Question 30.11. Which of the benzene ring hydrogen of toluene is the most crowded (or in the words of one student, “has the most shrubbery”)? Refer to your model of toluene to help you through this discussion. The para and meta hydrogens are each flanked by two hydrogens. The ortho hydrogens are flanked by a hydrogen atom and a methyl group. Hydrogen atoms are small and offer no significant hindrance as the electrophile approaches. However, a methyl group is larger than a hydrogen atom, and thus offers some steric hindrance to approach by the electrophile. Therefore we can conclude, based just on steric hindrance, that attack at the meta and para positions is favored over attack at the ortho position. This can be seen in the following space filling model. Ortho attack more hindered Meta attack less hindered Para attack less hindered We now have two factors that might influence the EAS product ratio. Based upon your organic chemistry experience so far, neither of these _________________________________________________________ Electrophilic Aromatic Substitution 15 factors is obviously dominant, so any conclusion must consider them both. Meta attack appears most favorable because there are two meta hydrogens and no steric hindrance. Whether attack at the ortho or para position is more favorable is a toss-up. The para position is less hindered but there is only one para hydrogen. The methyl group hinders the ortho position but there are two ortho hydrogens. However, we have yet to consider how the details of the reaction mechanism might also influence the product distribution. Think Ahead Question 30.12 Write the complete mechanism, including all significant resonance contributors, for the three products formed when toluene is reacted with Br2 and Fe. How do the differences in these mechanisms influence the product distribution? Answer: Contrasting the three mechanisms requires us to focus on their differences. Mechanism details that are the same for all three bromotoluene isomers will not help us here. Formation of the Br2FeBr3 complex does not vary, so we can ignore it for this discussion. The next portion of the mechanism, electrophilic attack on the benzene ring leads to three different arenium ions, so lets start our exploration at that point. Ortho attack: CH3 δ+ Br Br CH3 FeBr3 CH3 H CH3 H H Br Br Br Meta attack: CH3 δ+ Br Br CH3 CH3 CH3 FeBr3 Br Br Br H H H Para attack: CH3 CH3 δ+ Br Br FeBr3 H Br CH3 CH3 H Br H Br _________________________________________________________ Electrophilic Aromatic Substitution 16 Recall that in general, the formation of the more stable mechanism intermediate (the arenium ion, in this case) has a lower energy of activation, a greater reaction rate for the mechanism pathway, and therefore a greater abundance of the corresponding reaction product. (Review section 20.xx if this concept isn’t fresh in your mind.) So which arenium ion is more stable? The arenium ion derived from ortho attack has three significant resonance contributors, two of which are secondary carbocations and one of which is tertiary. Meta attack gives an arenium ion that also has three resonance contributors, all of which are secondary carbocations. Para attack, like ortho attack, also gives an arenium ion with three significant resonance contributors, two of which are secondary carbocations and one of which is tertiary. Therefore the arenium ion from meta attack is less stable than the arenium ion from ortho or para attack. Based only on arenium ion stability, we predict that the ortho and para products will be formed in greater amounts than the meta product. So let’s summarize the predictions based on the three factors discussed above. Factor Steric hindrance Probability Arenium ion stability Product Ratio Prediction meta, para > ortho ortho, meta > para ortho, para > meta These factors are not synergistic, and no obvious conclusion can be drawn, unless we can decide which factor is dominant. To do this we need some actual experimental data, which is given in Table 30.1. (The mechanisms for each of these reactions are discussed later on in this topic.) _________________________________________________________ Electrophilic Aromatic Substitution 17 Remember that a methyl group stabilizes an adjacent carbocation by electron donation. Review section 20.xx on carbocation stability if necessary. Table 30.1: Toluene EAS Product Ratios CH3 CH3 CH3 CH3 X EAS X X Ortho Meta Para 60 59 <1 4 40 37 57 3 40 42 19 39 9 1 89 Br2, Fe (X = Br) Section 30.4A Cl2, Fe (X = Cl) Section 30.4B HNO3, H2SO4 (X = NO2) Section 30.9 CH3CH2Cl, AlCl3 (X = CH2CH3) Section 30.11 O O PhCCl, AlCl3 (X = PhC ) Section 30.12 This data is most consistent with arenium ion stability as the dominant factor. Furthermore, probability carries more weight than steric effects, unless the steric effects are severe, as illustrated by the data in Table 30.2. This order of importance applies to the vast majority of EAS reactions. Table 30.2 Ortho : Para Product Ratios for Nitration of Alkylbenzenes R R R NO2 HNO3 H2SO4 NO2 R = CH3 R = CH2CH3 R = C(CH3)3 59 50 18 41 50 82 _________________________________________________________ Electrophilic Aromatic Substitution 18 In General... Relative Importance of Factors Influencing Regioselectivity in EAS Reactions: Arenium ion stability > number of hydrogens > steric effects The methyl group controls the regioselectivity of toluene EAS reactions. It serves the same role as the director in a movie, telling the actors where to go, so we call the methyl group an ortho/para director. The methyl group exerts this directing effect because the arenium ions from electrophilic attack to the ortho and para positions are more stable than the arenium ion that comes from meta attack. There are many different substituents that a benzene ring might have. Can we make a generalization about which ones are ortho/para directors? Think Ahead Question 30.13 A methyl group is an ortho/para director because it stabilizes an adjacent carbocation. What other benzene ring substituents might also be ortho/para directors? It may be useful to review the list of functional groups in section 1.xx and discussion of carbocation stability in section 24.xx. Answer: A methyl group exerts its directing effect by releasing electron density to an adjacent carbocation, thereby increasing arenium ion stability. Therefore any atom or group that can release electron density should also be an ortho/para director. What features must the director have in order to release electron density? We have already visited this issue when we explored carbocation stability (section 24.xx). We found that alkyl groups release electron density by inductive effects. We also found that carbocations enjoy significant resonance stabilization from adjacent carbon-carbon pi bonds (alkenes, alkynes and aromatic rings) and atoms with lone pairs (oxygen, nitrogen and the halogens being most important). Therefore we predict that alkyl groups, and any group that has a pi bond or lone pair adjacent to the benzene ring will be an ortho/para director. _________________________________________________________ Electrophilic Aromatic Substitution 19 Ortho/para director: In EAS, a benzene ring substituent that causes attack at the ortho and para positions of the benzene ring. R Alkyl groups stabilize arenium ions by inductive effects. Pi bonds stabilize arenium ions by resonance. X X Lone pairs stabilize arenium ions by resonance. Of course all of these groups are not as equally effective or influential. The effectiveness of resonance stabilization varies with different atoms and groups. For example, atoms that are more electronegative or groups with strong inductive electron-withdrawing effects provide less resonance stabilization. Table 30.3 summarizes the relative strength of various ortho/para directors. As you examine the substituents in Table 30.3 try to understand how each entry exerts its ortho/para directing effect. _________________________________________________________ Electrophilic Aromatic Substitution 20 Table 30.3 Relative Influence of Ortho/Para Directors Substituent Directing Strength H Directing Cause Lone pair resonance strongly activating N H R N H R N R OH OR - O N H R O O R Ar H weakly activating R Inductive effects Aromatic ring resonance None (reference point) In General Aromatic ring substituents that release electrons, either through resonance or inductive effects, are ortho/para directors. Concept Focus Question 30.5 Draw all of the important resonance contributors for the arenium ions formed from each of these molecules when reacted with Br2 and Fe. Without referring to Table 30.3 decide whether of not the substituent is an ortho/para director. OH (a) H N(CH3)2 (b) (c) N O H3C _________________________________________________________ Electrophilic Aromatic Substitution 21 The “Ar” abbreviation means “any aromatic ring” (not “argon”!). O N(CH3)3 (d) (e) N H H3C Concept Focus Question 30.6 Briefly explain. (a) NH2 is a stronger director than OH. (b) An amine is a stronger director than an amide. (c) NH2 is a stronger than N(CH3)2 (Hint: Examine models of the arenium ions.) Concept Focus Question 30.7 Suggest the major product of each reaction. (a) OCH3 Br2, Fe (d) CH3O O Cl2, FeCl3 (e) H3C CH3 Cl2, Fe CH3CH2 (b) C(CH3)3 N H (c) I2, HgO B. Activating Effects We have discussed how electron-releasing substituents influence the regioselectivity of an EAS reaction: they are ortho/para directors. But how do they influence the rate? Think Ahead Question 30.14 Rank these molecules in order of increasing reaction rate with Br2 and Fe: benzene, toluene and phenol. Answer: Remember that for a reaction mechanism with more than one step, it is the step with the highest energy of activation that has the greatest influence on the reaction rate. This is the rate-determining step (rds). It is the rds that controls the overall reaction rate, so we need only consider this step when exploring the effects of some variable (such as the identity of benzene ring substituents) on reaction rate. _________________________________________________________ Electrophilic Aromatic Substitution 22 Cl2, Fe So what is the rds for EAS? It is electrophilic attack on the benzene ring (section 30.3). In this mechanism step, the benzene ring (including its substituents) is the nucleophile, and an arenium ion is produced. How can a substituent influence nucleophilicity? As we have in many cases before (sections 21.xx and 22.xx, for example) enhancing the nucleophile’s electron density makes the reaction faster. Therefore any substituents on the benzene ring that release electron density enhance the reaction rate. This electron release might occur by inductive effects (methyl and other alkyl groups do this) or by resonance (groups like OH with an adjacent lone pair or pi bond). Therefore toluene and phenol are more nucleophilic than benzene, so toluene and phenol undergo nucleophilic attack more rapidly than benzene. Furthermore, resonance is generally more effective at increasing nucleophilicity than inductive effects (sections 21.xx and 22.xx), so phenol undergoes electrophilic attack faster than toluene. The electrostatic potential maps are useful to visualize a substituent’s influence on benzene ring electron density. The center of the benzene ring is red, indicating this is an area of lower electron density. In toluene, where the methyl group is a weak electron donor (inductive effect), the red area is diminished slightly. In phenol, where the hydroxyl is a strong electron donor (resonance), the red area is significantly diminished. Molecule Electrostatic Potential Map Comments H H H H H A hydrogen atom does not release electron density into the ring. H CH3 H H H H An alkyl group weakly releases electron density by inductive effects. H _________________________________________________________ Electrophilic Aromatic Substitution 23 OH H H H H Lone pairs strongly release electron density by resonance. H How can a substituent influence arenium ion stability? We have already explored this point in section 30.5A. The methyl group of toluene enhances arenium ion stability by electron release through an inductive effect. The hydroxyl group of phenol enhances arenium ion stability by resonance. The formation of an arenium ion that is more stable has a lower activation energy and faster reaction rate. Therefore toluene and phenol accept an electrophile faster than benzene. Once again, resonance is generally more effective for carbocation stabilization than an inductive effect, so phenols undergo electrophilic attack faster than toluene. So to answer Think Ahead Question 30.14, the EAS reaction rates are: phenol (fastest) > toluene > benzene (slowest). A c t i v a t o r : In EAS, an aromatic ring substituent that enhances nucleophilicity and arenium ion stability, thereby accelerating the reaction. It is no coincidence that our exploration of substituent effects on nucleophilicity and arenium ion stability reached the same conclusion. This is because the same factor – electron density – is at work in both cases. Electron-releasing substituents lead to faster reactions, and are called activators. We also predict that activators are also ortho/para directors, because electron density influences reaction rate and regioselectivity in the same way. Experimental data agrees with this prediction (although there is one important exception, discussed section 30.7), so we can make a general statement. In General In EAS reactions, substituents that release electron density into the benzene ring by resonance or inductive effects are ortho/para directors and activators. (For an important exception to this generalization, see section 30.7). If the benzene ring has sufficient electron density, because the activator (or combination of activators) is strong enough, then a weaker electrophile can be used. For example, toluene has a weak activator (CH3), and is inert to Br2 without FeBr3 or I2 without HgO. On the other hand, phenol has a moderate activator (OH), so it has enough electron density to react with Br2 or I2 without the need for additional reactants. In fact, phenol is so strongly activated towards _________________________________________________________ Electrophilic Aromatic Substitution 24 EAS that bromination does not stop until all of the ortho and para positions have been brominated. Aniline (PhNH2) behaves similarly. CH3 CH3 CH3 Br Br2, FeBr3 + Fails without FeBr3 Br OH OH excess Br2 Br Br No FeBr3 necessary Br Concept Focus Question 30.8 In each pair of molecules, select the one that reacts fastest with Br2/Fe. Draw the products of the faster reaction. (a) Toluene or benzene (c) Toluene or p-xylene (b) Toluene or aniline (d) Phenol or anisole Concept Focus Question 30.9 Write a complete mechanism for the reaction of phenol with excess Br2 to give 2,4,6-tribromophenol. 30.6 Substituent Effects: Electron-Withdrawing Groups In section 30.5 we learned that electron-releasing groups are ortho/para directors and activators. Not all benzene ring substituents are electronreleasing groups, however. What is the effect of electron-withdrawing groups on EAS regioselectivity and reaction rate? Think Ahead Question 30.15 Predict the EAS product(s) when nitrobenzene is reacted with Br2 and Fe. Is this reaction faster or slower than the same reaction with benzene? Hint: Consider the Lewis structure and resonance of the nitro group. Answer: We learned in the previous section that a ring substituent controls EAS reaction rate and regioselectivity by its influence on the electron density of the benzene ring. This electron density controls the nucleophilicity of the ring and the stability of the arenium ion. _________________________________________________________ Electrophilic Aromatic Substitution 25 Review section 1.xx if you have trouble figuring out the Lewis structure of the nitro group. The nitro group is not a functional group we have encountered frequently, so let us begin our search to answer Think Ahead Question 30.15 by reviewing its structure. The Lewis structure shows the nitrogen atom is bonded to the benzene ring and two oxygen atoms. The nitro group has two significant resonance contributors, each with a +1 charge on the nitrogen and a -1 charge on one oxygen. O O N N O D e a c t i v a t o r : In EAS, an aromatic ring substituent that decreases nucleophilicity and arenium ion stability, thereby slowing the reaction. O What does this Lewis structure suggest about the nitro group’s effect on the electron density of the benzene ring? A nitrogen atom with a formal positive charge is highly electronegative (more so than a nitrogen without a formal charge), so it is drawing electron density from the ring toward itself. This effect is amplified by the inductive effect (section 21.xx) of the two oxygen atoms. Taken together these observations suggest the nitro group to be a powerful electronwithdrawing group. It reduces benzene ring nucleophilicity and decreases arenium ion stability, causing a slower EAS reaction. Therefore the nitro group is a deactivator. By extension, any electronwithdrawing group is also a deactivator. What about the regioselectivity for EAS bromination of nitrobenzene? As before, we need to consider the arenium ions derived from attack at the ortho, meta and para positions. Are all of these contributors important? Do all of the contributors increase arenium ion stability? Ortho attack: O N O O δ+ Br Br FeBr3 N O O N O O Br O H H H N Br Br Unimportant contributor _________________________________________________________ Electrophilic Aromatic Substitution 26 Meta attack: O O N O δ+ Br Br N O O N O O N O FeBr3 Br Br Br H H H Para attack: O N O O δ+ Br Br FeBr3 H N O Br O N O H Br Unimportant contributor O H N O Br The arenium ion derived from ortho attack has three resonance contributors, all of which are secondary carbocations. However the third contributor is not very significant because it has a formal positive charge on adjacent atoms. So this arenium ion is better described as having just two resonance contributors. (We can also say that it is not significant because the strong electron-withdrawing group causes significant destabilization of the adjacent electron-poor carbocation.) The arenium ion derived from para attack also has just two significant resonance contributors for the same reason. In contrast, none of the resonance contributors for the arenium ion from meta attack have a problem with adjacent positive charges, so all three are significant. In summary, the arenium ions from ortho and para attack each have two significant resonance contributors whereas the arenium ion from meta attack has three. Therefore meta attack leads to a more stable arenium ion, so it is the preferred pathway. In other words, the nitro group is a meta director. Think Ahead Question 30.16 A nitro group is a meta director because it destabilizes an adjacent carbocation. What other benzene ring substituents might also be meta directors? It may be useful to review the list of functional groups in section 1.xx and carbocation stability in section 24.xx. The nitro group exerts its meta directing effect because it is electron withdrawing. Based on this we can predict that any electronwithdrawing substituent will be a meta director. Experimental data shows this prediction to be true. Table 30.4 lists some commonly encountered meta directors. Like ortho/para directors, we can also rank meta directors according to the strength of the influence on the rate of _________________________________________________________ Electrophilic Aromatic Substitution 27 Meta director: In EAS, a benzene ring substituent that causes attack at the meta positions of the benzene ring. EAS reactions. However, because the interplay of electronegativity, inductive effects and other factors, it is not always readily apparent why one meta director should be weaker or stronger than another. In General In EAS reactions, substituents that withdraw electron density from the benzene ring are meta directors and deactivators. Table 30.4 EAS Meta Directors Substituent Directing Strength N O NH2R, NH3, NHR2, NR3 CF3 strongly deactivating O O S OH O C N O Cl O X O R O weakly deactivating X = OH, OR, NH2, NHR, NR2 H _________________________________________________________ Electrophilic Aromatic Substitution 28 CAUTION!!! You may think that benzene ring substituents that feature an atom of high electronegativity directly attached to the ring (such as OH or F) are meta directors because they destabilize an adjacent carbocation by an electron withdrawing inductive effect. However, these atoms usually have one or lone pairs, which stabilize an adjacent carbocation by resonance, causing ortho/para direction. Using our usual assumption that resonance dominates other factors suggests (correctly so) that the substituents in question are all ortho/para directors. X X has high electronegativity: Inductive electron-withdrawing group Less influential directing effect X X has lone pair: Resonance electron-donating group More influential directing effect Concept Focus Question 30.10 A methyl group is an ortho/para director but a trifluoromethyl group (CF3) is a meta director. Explain. Illustrate this point by writing the reaction products formed when toluene and trifluoromethylbenzene are treated with Br2 and Fe. Concept Focus Question 30.11 (a) Explain why all functional groups in which a carbonyl is directly bonded to the benzene ring (such as PhCO2H) are meta directors. (b) Explain why an acid chloride is a stronger meta director than a ketone. Concept Focus Question 30.12 Write the major product of each reaction. (a) NO2 (b) CO2H Br2, Fe Cl2, FeCl3 (c) CF3 (d) I2, HgO NO2 Br2, Fe O2N _________________________________________________________ Electrophilic Aromatic Substitution 29 30.7 Substituent Effects: The Halogens You may have noticed that we have avoided discussing the directing and activating effects of halogens, even though these are common and important benzene ring substituents. This is because their behavior is a bit quirky, as we will see in the following section. Think Ahead Question 30.17 Based upon what we have already learned about EAS directing and activating effects, write the reaction mechanism (including all arenium ion resonance contributors) and major product(s) for the reaction of fluorobenzene with Cl2 and Fe. F Cl2, Fe ??? Answer: As we have seen so many times before EAS regioselectivity is controlled by arenium ion stability. Fluorine has three lone pairs that can stabilize an adjacent carbocation by resonance (just like oxygen and nitrogen) so this suggests it is an ortho/para director. The lone pairs provide an extra, very significant resonance contributor in which all atoms have full octets. F F δ+ Cl Cl FeCl3 H F F Cl H Cl H F Cl H Cl Most important contributor On the other hand, fluorine’s high electronegativity destabilizes an adjacent carbocation, suggesting it to be a meta director. However (as we were reminded in the previous Caution box) resonance usually outweighs inductive effects, so we predict the net effect of fluorine to be an ortho/para director. We also predict fluorine to be an activator for the same reason it is an ortho/para director. The lone pairs increase the nucleophilicity of the benzene ring and provide extra stability for the arenium ion. This reduces the energy of activation for electrophilic attack, and results in a faster reaction. _________________________________________________________ Electrophilic Aromatic Substitution 30 So is our prediction accurate? To test it, we turn to experimental data, which is shown below. F F Cl2, Fe F Cl + F + Cl 30% 1% Cl 69% The data reveals that our regioselectivity prediction was accurate, and fluorine is an ortho/para director. However, our rate prediction does not agree with the experimental data. We predicted reaction of fluorobenzene to be faster than similar reaction of benzene, but the opposite is true. If the experimental rate for chlorination of benzene is taken as 1.0 then the rate for chlorination of fluorobenzene is 0.74, a little bit slower. (In other words, fluorobenzene is almost as reactive as benzene in EAS, despite the presence of the most electronegative atom in the periodic table!) A similar rate effect is observed for EAS reactions with chlorobenzene, bromobenzene and iodobenzene. Their rates are all slower than benzene itself, even though all halogens have lone pairs for resonance donation. The explanation for the unexpected EAS behavior of halogens (weakly deactivating ortho/para directors) has been debated, but the general agreement is that there is a subtle balance between their inductive electron-withdrawing properties and their resonance electron-donating properties, resulting in the observed reactivity. In General In EAS reactions, a halogen benzene ring substituent is a weakly deactivating ortho/para director. _________________________________________________________ Electrophilic Aromatic Substitution 31 CAUTION!!! The regioselectivity of an EAS reaction is controlled by the substituent(s) that are attached to the benzene ring at the start of the reaction, and not the incoming electrophile (the group that is added to the ring in the course of the reaction). For example, nitro is a meta director and bromine is an ortho/para director. Bromination of nitrobenzene gives m -bromonitrobenzene whereas nitration of bromobenzene gives a mixture of o- and p-bromonitrobenzene. NO2 NO2 Meta director Br2, Fe Br Br Br aq. HNO3 ortho/para director NO2 aq. H2SO4 Br + NO2 Concept Focus Question 30.13 Rank these molecules by their reaction rates in a typical EAS reaction: benzene, toluene, fluorobenzene, and p-difluorobenzene. Concept Focus Question 30.14 Write the major products of these reactions. F Br Br2, Fe (a) Cl (b) I2, HNO3 (c) F Cl2, FeCl3 Br2, Fe (d) F 30.8 Substituent Effects: Effect of Multiple Substituents Most of the EAS cases we have examined up to this point have involved a benzene ring bearing a single substituent. In some of the Concept Focus Questions we have explored cases with two equal _________________________________________________________ Electrophilic Aromatic Substitution 32 substituents. In these cases there was little question as to which product is formed because the substituent directing effects are cooperative. What happens if the directing effects are not cooperative? Lets look at some experimental data then derive a general statement. Think Ahead Question 30.18 Based on the reactions shown below, formulate a general rule concerning the regioselectivity of EAS on benzene rings bearing multiple substituents. Only the major product is shown in each case. OCH3 OCH3 Br Br2 Reaction A: CH3 CH3 OCH3 OCH3 Br2, Fe Reaction B: Br NO2 CO2H Reaction C: NO2 Br2, Fe CO2H Br NO2 NO2 Answer: Lets analyze each case individually and then try to find a pattern. Check the substituent directing effect lists (Tables 30.3 and 30.4) as needed, but only after you have tried to deduce the effects you have forgotten. Reaction A: Methoxy is a strongly activating ortho/para director and methyl is a weakly activating ortho/para director. (This benzene is so electron rich that Br2 does not have to be activated with FeBr3.) The product of this reaction tells us that the stronger ortho/para director has more influence than the weaker ortho/para director. stronger o/p director OCH3 OCH3 Br Br2 CH3 weaker o/p director CH3 _________________________________________________________ Electrophilic Aromatic Substitution 33 Reaction B: Methoxy is an activating ortho/para director and nitro is a deactivating meta director. The product of this reaction tells us that the activating ortho/para director wins out over the deactivating meta director. The relative strength of the directing effects even overrides the greater steric hindrance of the methoxy group. strong o/p director OCH3 OCH3 Br2, Fe Br NO2 NO2 strong m director Reaction C: Nitro is a strongly deactivating meta director and a carboxylic acid is a weakly deactivating meta director. The product of this reaction tells us that the weaker deactivating group controls the regioselectivity. weaker m director CO2H Br2, Fe NO2 Br CO2H NO2 stronger m director What is the general ranking of the relative influence of two substituents? In General Stronger activators dominate over weaker activators (reaction A). Weaker deactivators dominate over stronger deactivators (reaction C). Activators dominate over deactivators (reaction B). Many reactions obey this general ranking, regardless of the number of substituents, although there are exceptions. When the substituents are very similar in activating or deactivating strength, such as OCH3 versus NHCOCH3, all products are formed in roughly equal amounts, although steric effects can influence this ratio. Concept Focus Question 30.15 Rank each set of substituents in order of decreasing influence on EAS reactions. (a) -OH, -CH2CH3, -NO2 (b) -OH, -OCH3, -F (c) -F, -CF3, -NO2 _________________________________________________________ Electrophilic Aromatic Substitution 34 Concept Focus Question 30.16 Give the major product of each reaction. CH2CH3 Cl Br2, Fe Br2 (a) (c) OH NH2 NO2 CO2H Cl2, Fe (b) (d) CH3 Cl2, Fe CO2CH3 O Up to this point we have only discussed EAS reactions that introduce halogens. There are many other substituents that we can introduce using EAS. All of these reactions follow the same general pattern we have seen so far: an electrophile (Elec) attacks the aromatic ring, ultimately replacing one of its hydrogens (Ar-H becomes Ar-Elec). For each new EAS reaction we explore we will deduce the nature of the electrophile by examining EAS reactions that are (or have been) important on an industrial scale. 30.9 Nitration Think Ahead Question 30.19 TNT (2,4,6-trinitrotoluene) was once prepared by reacting toluene with an aqueous mixture of nitric and sulfuric acids until three nitro groups were added. (TNT is no longer manufactured on a large scale.) What is the electrophile in this reaction? What is the mechanism for the conversion of toluene into p-nitrotoluene? Hint: You will need the correct Lewis structure for nitric acid. CH3 CH3 aq. HNO3 + aq. H2SO4 NO2 CH3 CH3 NO2 CH3 NO2 aq. HNO3 aq. H2SO4 aq. HNO3 NO2 O2N aq. H2SO4 NO2 NO2 2,4,6-trinitrotoluene (TNT) _________________________________________________________ Electrophilic Aromatic Substitution 35 Answer: Let us draw on what we already know about the EAS mechanism. In the case of halogenation, the electrophile is a naked halogen atom such (e.g., Br+) or a halogen cation bearing a leaving group (e.g., the Br2-FeBr3 complex). Br comes from + Br New bond forms here Nitronium cation: O N O Applying the same idea to nitration, the electrophile is +NO2 (the nitronium cation) or NO2 bearing a leaving group. Experimental evidence supports the nitronium cation as the actual electrophile. NO2 comes from + NO2 How is +NO2 formed? Nitric acid could lose an HO group, but hydroxide is not a leaving group under these conditions. The OH can be converted to water (a moderate leaving group) by protonation, a feat easily achieved by H3O+ (from aqueous H2SO4). Once generated, + NO2 achieves EAS by the same general mechanism we developed in section 30.3. Mechanism: Electrophilic Aromatic Nitration O H2O H HO H2O O N N O Protonation makes HO a better LG +NO 2 Nitronium ion (an electrophile) O Water leaves CH3 CH3 O O O N N O adds to give most stable arenium ion O2N H CH3 OH2 NO2 Deprotonation restores aromaticity Why does nitric acid accept the proton at the OH oxygen instead of the oxygen with a negative formal charge? Protonation of the OH oxygen _________________________________________________________ Electrophilic Aromatic Substitution 36 does not cause a significant loss of resonance whereas protonation of the negatively charged oxygen does. EAS nitration is a fairly general reaction but it does have some limitations. Nitric acid oxidizes primary and secondary amines, so the reaction gives other products if the ring has an –NH2 or –NHR group. Tertiary amines (-NR2) are not oxidized by nitric acid. Another reagent that has been used is nitronium tetrafluoroborate (+NO2 -BF4). This is a milder, much less acidic way to achieve EAS nitration. Concept Focus Question 30.17 Suggest the major product(s) of these reactions. CH3 OCH3 (a) aq. HNO3 Ph O F3C aq. HNO3 CH3 NO2 -BF4 (c) aq. H2SO4 (b) + CF3 (d) aq. HNO3 aq. H2SO4 aq. H2SO4 30.10 Sulfonation Benzene sulfonic acids (Ar-SO3H) are present in several classes of organic molecules that are manufactured on multi-ton scales, including dyes and synthetic detergents. The sulfonic acid group can be introduced onto the benzene ring by EAS, illustrated here as one step in the synthesis of a detergent. R SO3 H2SO4 R = linear alkyl chain, e.g., C12H25 Sulfonic acid: O S OH O O R S OH + R O HO3S Think Ahead Question 30.20 What is the electrophile in the EAS sulfonation reaction? What is the overall mechanism? Answer: As in the previous section, we deduced the electrophile by working backwards. The new substituent is SO3H so the electrophile _________________________________________________________ Electrophilic Aromatic Substitution 37 A mixture of SO3 and H2SO4 is called fuming sulfuric acid or oleum. might be +SO3H. Working out the correct Lewis structure for +SO3H suggests it might come from protonation of SO3 by H2SO4. O O S comes from OH + S O O O + OH S O OH S comes from O + "H " O O Now we can write the complete mechanism for electrophilic aromatic sulfonation. Mechanism: Electrophilic Aromatic Sulfonation O O O S O H S OSO3H OH O O O S OH Protonation of SO3 makes it a stronger electrophile R R R O O S HO3S H OSO3H SO3H OH + SO3H adds to give most stable arenium ion Deprotonation restores aromaticity A slightly different mechanism is also possible in which SO3 itself is the electrophile. The sulfur atom carries a large δ+ charge due to the three highly electronegative oxygen atoms. If SO3 is the electrophile the mechanism varies only in the timing of the proton transfer steps (see Concept Focus Question 30.19 at the end of this section). Experimental evidence suggests that concentration, temperature, aromatic substrate, and other variables determine whether the actual electrophile is SO3 or +SO3H. In this textbook we will accept either mechanism for this reaction. (Other related electrophiles such as protonated sulfuric acid, H3SO4+, have also been implicated.) Unlike other EAS reactions, sulfonation is easily reversible, so the SO3H group can be replaced with a hydrogen atom. The _________________________________________________________ Electrophilic Aromatic Substitution 38 benzenesulfonic acid need only be heated with dilute aqueous acid to remove the sulfonic acid group. SO3, H2SO4 SO3H H3O+ So a sulfonic acid group is a removable meta director. Its addition and later removal can be a useful strategy to control regioselectivity of other EAS reactions, and to synthesize substituted benzene derivatives that may not be easily made by other routes. For example, consider the synthesis of m-dibromobenzene from benzene. Treatment of benzene with excess Br2 and Fe gives ortho- and para-dibromobenzene, but not the meta isomer. Br Br Br Br Fe, Br2 Fe, Br2 + Br However, bromination of benzenesulfonic acid followed by desulfonation gives the desired isomer. SO3H SO3 SO3H H3O+ Fe, Br2 H2SO4 Br Br Br Br Concept Focus Question 30.18 Give the major products of these reactions. (a) C(CH3)3 O (b) SO3 H2SO4 SO3 H2SO4 (c) (d) SO3H HO3S SO3 H2SO4 I H3O+ heat Concept Focus Question 30.19 Write the mechanism for the sulfonation reaction of Concept Focus Question 30.18(a), using SO3 (not +SO3H) as the electrophile. _________________________________________________________ Electrophilic Aromatic Substitution 39 Concept Focus Question 30.20 Write the mechanism for the desulfonation reaction of Concept Focus Question 30.18(d). 30.11 Friedel-Crafts Alkylation In sections 30.4, 30.9 and 30.10 we explored reactions for introducing halogens, nitro groups and sulfonic acid groups onto a benzene ring. Synthesis of complex molecules often involves building a more complex carbon skeleton, so can we use EAS to add alkyl groups to a benzene ring? Friedel-Crafts alkylation: An EAS reaction in which an alkyl carbocation or related structure adds an alkyl group to the aromatic ring. Think Ahead Question 30.21 In 1877, Charles Friedel and James Craft published a new reaction for making bonds between sp3 carbons and benzene ring carbons. This reaction came to be called the Friedel-Crafts alkylation. For example, reaction of tert-butyl chloride, benzene and aluminum chloride gives a mixture of ortho- and para-tert-butyltoluene. What is the electrophile in this EAS reaction? Write a complete mechanism for the EAS reaction. CH3 CH3 (CH3)3CCl CH3 C(CH3)3 + AlCl3 C(CH3)3 Answer: Using the same EAS analysis strategy we have used in the past several sections of this topic, we can disconnect the electrophile from the benzene ring, revealing it to be a tert-butyl carbocation. CH3 CH3 comes from + C(CH3)3 C(CH3)3 We have produced carbocations before by ionization of a carbonleaving group bond (SN1 and E1 reactions) or in some pi bond addition reactions. Either of these carbocation sources could also be used in the _________________________________________________________ Electrophilic Aromatic Substitution 40 EAS reaction. Historically, an SN1-like reaction was developed first, so that is where we shall start. The tert-butyl carbocation can be formed by ionization of tert-butyl chloride, but as we saw before (sections 25.xx and 27.xx) this is an energetically expensive process. The role of the aluminum chloride is to assist this ionization by first forming a Lewis acid-halogen bond (just as we saw in EAS bromination and chlorination, section 30.4). With these issues resolved we can write the mechanism. Mechanism: Friedel-Crafts Alkylation (CH3)3C Cl AlCl3 Lone pair from chlorine fills open octet of aluminum (CH3)3C Electrophile adds to benzene ring (CH3)3C + ClAlCl3 AlCl3 Chlorine leaving group ability improved; bond ionizes CH3 CH3 CH3 C(CH3)3 Cl (H3C)3C H Cl AlCl3 C(CH3)3 Deprotonation restores aromaticity The Friedel-Crafts reaction can be used to add a wide variety of alkyl groups to a benzene ring, but the mechanism cannot be exactly the same for all cases. Think Ahead Question 30.22 Write the reaction and mechanism for the conversion of cumene (isopropylbenzene) into p-methyl isopropylbenzene by a Friedel-Crafts reaction. Answer: Addition of a methyl group suggests the alkyl halide component of the reaction is CH3Cl. Thus the reaction is: CH3Cl AlCl3 CH3 _________________________________________________________ Electrophilic Aromatic Substitution 41 What is the electrophile? Methyl chloride reacts with AlCl3 to form a CH3Cl-AlCl3 complex. Ionization of this complex is difficult because it would form a very unstable methyl carbocation. This suggests the electrophile must be something other than a carbocation. Since the CH3Cl-AlCl3 complex has a leaving group, it can fulfill this role, as shown in the following mechanism. H3C Cl H3C AlCl3 Cl X AlCl3 H3C + ClAlCl3 CH3+ too unstable; no ionization H3C Incipient carbocation: A molecule in which carbon bears a large _+ charge and a good leaving group. Behaves in most ways like a carbocation. ClAlCl3 H3C H Cl AlCl3 CH3 The carbon-chlorine bond of the CH3Cl-AlCl3 complex is very weak and the carbon bears a large δ+ charge, more so than the carbon of CH3Cl itself. It is as if the carbon-chlorine bond is almost completely broken and the carbon is almost (but not quite yet) a carbocation. This is called an incipient carbocation. Because the carbon bearing the leaving group is secondary, the electrophile that attacks the benzene ring could be either a carbocation or an incipient carbocation. An incipient carbocation behaves very much like a normal “naked” carbocation. For example, it is prone to rearrangement as shown in the following example. CH3CH2CH2Cl AlCl3 You might ask why the Friedel-Crafts alkylation reaction of Think Ahead Question 30.21 involving (CH3)3C+ did not avoid the energetically expensive carbon-chlorine bond ionization and instead used the (CH3)3CCl-AlCl3 complex directly as the electrophile. The answer lies in steric effects: the carbon bearing the chlorine is tertiary, and thus too hindered to undergo nucleophilic attack. δ+ (CH3)3C Cl AlCl3 X No nucleophilic attack at 3o carbon _________________________________________________________ Electrophilic Aromatic Substitution 42 In General The electrophile in a Friedel-Crafts alkylation reaction can be a carbocation or an incipient carbocation (RCl-AlCl3 complex). The more stable the carbocation is, the greater chance that RCl-AlCl3 complex ionizes before attacking the benzene ring. When R is methyl or 1o the electrophile is the complex. When R is 2o either the complex or the carbocation can be the electrophile. When R is 3o the electrophile is most probably the carbocation. (This is analogous to the choice of SN2 versus SN1 mechanism for an ionic substitution reaction, section 25.xx.) Here are a few limitations of the Friedel-Crafts alkylation reaction. • The reaction fails if the benzene ring is too electron deficient due to the presence of one or more strong deactivating groups. (Several weak deactivating groups could combine to have a similar effect.) For example, the reaction does not occur if the benzene ring has a nitro, sulfonic acid, ketone or aldehyde substituent. This reduced reactivity is a combination of decreased benzene ring nucleophilicity plus reduced arenium ion stability. For this reason nitrobenzene is sometimes used as a solvent for Friedel-Crafts reactions. This problem can be resolved in some cases by running the reaction with an electron-rich benzene ring, then changing the substituents in subsequent reactions. • The product of a Friedel-Crafts alkylation is more activated that the starting material due to the presence of an additional electron-donating alkyl group. Thus the reaction product may alkylate faster than the starting material, and multiple alkylations can result. This problem can be addressed by using a large excess of the starting material, so that the electrophile has a great chance of colliding with a molecule of starting material before it finds a molecule of product. This problem can be avoided with the Friedel-Crafts acylation (section 30.12). CH3 CH3Cl + AlCl3 Major product only when benzene is used in large excess dimethylbenzenes + trimethylbenzenes + etc. Can be significant products if benzene is not in large excess _________________________________________________________ Electrophilic Aromatic Substitution 43 Most any alkyl halide can be used, including fluorides, chlorides, bromides and iodides. However aryl or vinyl halides cannot be used because the intermediate carbocations are too unstable to form and direct “SN2” attack at an sp2 carbon is not possible (section 22.xx). • Concept Focus Question 30.21 Write the major organic product(s) of these reactions. Assume that a large excess of the aromatic starting material is used in each case. (a) CH3CH2Cl (d) AlCl3 (b) O AlCl3 (e) CH3Cl ClCH2CPh3 CH3Cl AlCl3 AlCl3 O (c) CH3(CH2)3Cl AlCl3 Concept Focus Question 30.22 Write a mechanism for the reaction of Concept Focus Question 30.21(c). In the previous section we learned that in some Friedel-Crafts reactions, the electrophile is a carbocation. Does this suggest that any carbocation source can be used to alkylate a benzene ring? Think Ahead Question 30.23 Give a mechanism for the reaction of benzene and propene in the presence of sulfuric acid to give cumene. CH3CH=CH2 H2SO4 Answer: This appears to be an EAS reaction similar to a Friedel-Crafts alkylation because an alkyl group has replaced a benzene ring hydrogen. However, the lack of alkyl halide and AlCl3 suggest the mechanism is something different. So we start the mechanism analysis by disconnecting the alkyl group to determine the electrophile. _________________________________________________________ Electrophilic Aromatic Substitution 44 CH3 comes from and C H CH3 Aha! We know how to generate carbocations from alkenes by protonation (sections 24.xx and 28.xx). Protonation of propene by sulfuric acid gives a 2-propyl carbocation. So now we can write the mechanism. H OSO3H H OSO3H The use of only a small (catalytic) amount of H2SO4 and a large excess of benzene helps ensure the carbocation is captured by the benzene ring and not by –OSO3H. Liquid HF or other specialized acid catalysts can also be employed. Other carbocation sources such as alcohols are also useful. For example, 2-propanol could be used instead of propene in the previous reaction. Despite their limitations, the Friedel-Crafts alkylation reactions we have explored in this section are key steps in the industrial syntheses of several important products. These include ethylbenzene (converted to styrene for polymers), cumene (converted into phenol and acetone), long-chain alkylbenzenes (converted into sodium alkylbenzene sulfonates for detergents) and short-chain alkylbenzenes (blended in gasoline for higher octane ratings). 30.12 Friedel-Crafts Acylation Friedel-Crafts reactions can also be used for acylation, the addition of a new carbonyl-containing substituent to a benzene ring. For example, the reaction of thioanisole (PhSCH3) with acetyl chloride and aluminum chloride was part of the industrial-scale synthesis of the nonsteroidal anti-inflammatory analgesic (pain killer) Vioxx. CH3S CH3 CH3S O O S O more steps Cl O O AlCl3 Vioxx O _________________________________________________________ Electrophilic Aromatic Substitution 45 Vioxx was a very profitable drug for Merck, netting over $1 billion in annual sales, until it was implicated in increased risk of heart disease and stroke, and voluntarily withdrawn from the market in later 2003. Think Ahead Question 30.24 Determine the electrophile that is formed in the reaction of thioanisole, acetyl chloride and aluminum chloride. Write the complete mechanism. Friedel-Crafts acylation: An EAS reaction in which an acylium ion adds a ketone group to the aromatic ring. Answer: This reaction bears a striking resemblance to the FriedelCrafts alkylation (section 30.11), but an acyl group is added instead of an alkyl group. In fact was discovered by the same chemists, so it is called the Friedel-Crafts acylation reaction. Because of its similarity to the alkylation reaction of the same name, we might assume the mechanism to be similar as well. Lets start by applying the same disconnection analysis as we did in section 30.11. comes from CH3S CH3S O Acylium ion: R-C≡O + and O The electrophile is a carbocation, just like in the Friedel-Craft alkylation. The carbocation is formed by Lewis acid-assisted ionization of the carbon-chlorine bond, just like the Friedel-Crafts alkylation reaction. How easy is this ionization? The carbocation has resonance stabilization, so the ionization occurs readily. The resultant carbocation is called an acylium ion, because of its structural similarity to an acyl group. The acylium ion resonance contributor with a triple bond may look strange, but we have seen plenty of structures with positively charged oxygens atoms having three bonds, such as H3O+. Recall also that the resonance contributor with the maximum number of full octets contributes most to the hybrid (section 5.xx). Now we can write the complete mechanism. _________________________________________________________ Electrophilic Aromatic Substitution 46 Mechanism: Friedel-Crafts Acylation O O O O C H3C Cl H3C AlCl3 Cl O O CH3S H Cl AlCl3 CH3 Acylium ion adds to form most stable arenium ion H3C Acylium ion resonance contributors CH3S CH3S C H3C Ionization of weakened carbon - chlorine bond Lone pair from chlorine fills open octet on aluminum H3C AlCl3 O CH3 Deprotonation restores aromaticity Note how the mechanism is very similar to the Friedel-Crafts alkylation. The Friedel-Crafts acylation reaction has fewer limitations than the corresponding alkylation reaction. The most significant limitation is the reaction fails when the benzene ring is too electron deficient due to the presence of one or more strong deactivating groups (or several weak deactivating groups could combine to have a similar effect), just like the Friedel-Crafts alkylation. The same restriction overcomes the multiple alkylation problem of the Friedel-Crafts alkylation reaction. Acylation adds a new carbonyl substituent, which serves as a deactivator and prevents further alkylation. For example, reaction of benzene with methyl chloride and aluminum chloride gives a mixture of products bearing one, two and possibly more methyl groups (unless a large excess of benzene is used). On the other hand, reaction of benzene with acetyl chloride and aluminum chloride stops after just one acetyl group has been introduced. _________________________________________________________ Electrophilic Aromatic Substitution 47 CH3 CH3 CH3 CH3 CH3Cl + AlCl3 + CH3 O + other products CH3 O Cl only product formed AlCl3 The Friedel-Crafts alkylation reaction may also involve carbocation rearrangements. This may or may not be a problem, depending upon the product that is desired. However, acylium ions do not rearrange (rearrangement would sacrifice significant resonance stabilization), so the carbon skeleton of the acid chloride starting material is retained in the aryl ketone product. Cl Incipient 1o carbocation rearranges to 3o carbocation AlCl3 O O Cl AlCl3 Acylium ion does not rearrange Concept Focus Question 30.23 Give the major organic product(s) when the following molecules are reacted with acetyl chloride and aluminum chloride. If no reaction occurs write “NR.” (a) Ethylbenzene (b) p-Methoxyisopropylbenzene (c) Nitrobenzene _________________________________________________________ Electrophilic Aromatic Substitution 48 30.13 The Clemmensen and Wolff-Kishner Reductions A Friedel-Crafts reaction is a very versatile method to make a new carbon-carbon bond to an aromatic ring. Friedel-Crafts acylation provides ketones. Friedel-Crafts alkylation allows addition of a new methyl group (ArH → ArCH3), as well as a bond to a new secondary carbon (ArH → ArCHR2) or tertiary carbon (ArH → ArCR3). However, because of carbocation rearrangements, making a bond to a primary carbon (ArH → ArCH2R) is not very efficient. How then might we achieve this very useful transformation? Think Ahead Question 30.26 What sequence of reactions can be used to convert benzene into butylbenzene? Answer: The conversion of benzene into butylbenzene (PhCH2CH2CH2CH3) requires formation of a new carbon-carbon bond between the benzene ring and a butyl group. We know that in general, a Friedel-Crafts reaction can be used to make this carbon-carbon bond. CH2CH2CH2CH3 comes from and CH2CH2CH2CH3 However, 1-chlorobutane is not an efficient source of the butyl carbocation because most of the 1o incipient carbocation rearranges to a more stable 2o carbocation. Cl + AlCl3 minor major How can we add a four-carbon chain without rearrangement? This calls for a Friedel-Crafts acylation with butyryl chloride. This reaction solves half the problem by constructing the correct carbon skeleton (Ph-C-C-C-C) but it creates a new problem: an undesired ketone group. If we had an effective method to convert the ketone to a O PhCH2R) this might be a useful way to get methylene group (PhCR around the Friedel-Crafts limitation. _________________________________________________________ Electrophilic Aromatic Substitution 49 Remember that a reduction reaction is one in which the number of bonds between carbon and elements less electronegative than carbon (usually hydrogen) is increased (section xx.xx). O O How to remove C=O? Cl AlCl3 As you may have guessed by now, there are at least a dozen reactions that have been specifically developed (or discovered by accident) for reduction of a ketone to a methylene group. (The same reactions also reduce an aldehyde to a methyl group.) Two of these reduction reactions are discussed here. A. Clemmensen Reduction Clemmensen reduction: Reduction of a ketone to a CH2 group (or aldehyde to CH3) with Zn/Hg and aqueous HCl. An amalgam is an alloy with mercury. Gold amalgam was once used to make gold feelings for teeth, until concerns about mercury toxicity caused this method to be abandoned. Reduction of a ketone or aldehyde with zinc in the presence of mercury (as an amalgam) and hydrochloric acid is called the Clemmensen reduction. (The Zn/Hg amalgam reacts with acid more slowly than pure Zn, so the slower Zn/carbonyl reaction can compete with the faster Zn/acid reaction.) The reaction is most efficient with aryl ketones, but other aldehydes and ketones can also be reduced. The mechanism of the reaction is not completely understood. The Clemmensen reduction solution to Think Ahead Question 30.25 is shown here. O O Cl AlCl3 Zn/Hg aq. HCl Clemmensen reduction requires strongly acidic conditions, which may not be tolerated by acid-sensitive molecules. For these cases we can use the Wolff-Kishner reduction. B. Wolff-Kishner Reduction Wolff-Fischer reduction: Reduction of a ketone to a CH2 group (or aldehyde to CH3) with hydrazine and KOH. The Wolff-Kishner reduction is an alternate method to convert a ketone into a methylene group. (This reaction can also be used to reduce an aldehyde to a methyl group.) It involves heating the carbonyl compound with hydrazine (H2NNH2) and strong base (KOH) in a protic solvent with a high boiling point (such as ethylene glycol, HOCH2CH2OH, bp 197oC). The Wolff-Kishner reduction solution to Think Ahead Question 30.25 is shown here. _________________________________________________________ Electrophilic Aromatic Substitution 50 O O H2NNH2 Cl AlCl3 KOH The mechanism for the Wolff-Kishner reduction is discussed in section 34.xx. Wolff-Kishner reduction requires strongly basic conditions, which may not be tolerated by base-sensitive molecules. For these cases we can use the Clemmensen reduction. The Wolff-Kishner reduction may not work if the ketone has too much steric hindrance. Concept Focus Question 30.24 Give the major organic product(s) of the following reactions. O (a) Zn/Hg aq. HCl O (b) H2NNH2 KOH H3C C(CH3)3 (c) O Cl , AlCl3 1. 2. Zn/Hg, aq. HCl 1. AlCl3 (d) 2. H2NNH2, KOH O Cl Concept Focus Question 30.25 Suggest a series of reactions to achieve each of these transformations. Select the most efficient route possible, using the least number of reactions. Avoid using only a minor product in the next step. (a) Benzene into 4-tert-butylacetophenone (b) Tert-butylbenzene into 5-tert-butyl-2-ethylnitrobenzene (c) Benzene into m-bromobutylbenzene _________________________________________________________ Electrophilic Aromatic Substitution 51 30.14 Diazo Coupling Diazo compound: Any molecule with the general formula R-N=N-R. Molecules containing a nitrogen-nitrogen double bond are called diazo compounds. If the diazo group links two aromatic rings, a highly conjugated molecule results. This conjugation causes the molecule to be strongly colored, often red, orange or yellow. For example, butter yellow (p-dimethylaminoazobenzene) has an intense yellow color. Its name comes from its old application for dyeing of white margarine to give it a more appealing, buttery yellow color. (For a more detailed discussion of how structure controls color, see section 17.xx.) N Azobenzene (orange-red) N (CH3)2N N N (CH3)2N SO3 Na N Methyl orange (orange) Butter yellow (yellow) N Lets explore the synthesis of butter yellow, using EAS chemistry. (We will examine a more complex case, the industrial scale synthesis of FD&C Red No. 40, in section 30.16, In The Real World.) Think Ahead Question 30.26 Butter yellow is synthesized by an electrophilic aromatic substitution reaction using N, N-dimethylaniline is the nucleophile. What is the electrophile? Answer: The diazo group may look alien to you, but no need to panic! We know an EAS reaction is involved, so we apply the same logic to determine the electrophile as we used previously (sections 30.9-30.12). (CH3)2N N N from nucleophile from electrophile comes from _________________________________________________________ Electrophilic Aromatic Substitution 52 and (CH3)2N N N N,N-Dimethylaniline the EAS nucleophile Benzenediazonium cation the EAS electrophile N N This analysis suggests the electrophile has a nitrogen-nitrogen triple bond bearing a formal positive charge. A molecule of the general structure RN≡N+ is called a diazonium cation. For the synthesis of butter yellow we need the benzenediazonium cation, PhN2+. (Of course the benzenediazonium cation is not a naked cation, but instead is accompanied by an anion, usually chloride ion. So diazonium compounds are best described as salts. However the anion is not particularly important in this process so we will ignore it.) Diazonium cations are unstable, because nitrogen gas is an exceptionally good leaving group. (Their high instability may lead to an explosion!) Diazonium salts are not the kinds of things we can make and keep around in a bottle until needed. Instead, diazonium salts are generally made when needed and used right away. Even though they are unstable, diazonium salts are easy to make: Reaction of a primary amine (such as aniline) with sodium nitrite (NaNO2) in the presence of aqueous acid does the trick. The conversion of an amine to a diazonium salt is called diazotization. Think Ahead Question 30.27 Work out the reaction mechanism for diazotization of aniline (i.e., its conversion into benzenediazonium chloride) with NaNO2 and aqueous HCl. Hint #1: The mechanism involves NO+, the nitrosonium cation. Hint #2: The mechanism is one of the longest you have seen so far, but it involves the kinds of steps you have seen before, such as proton transfers, leaving group departure and resonance. Make logical decisions based upon normal rules of molecular structure, while keeping in mind the structure of final product, and you should have little trouble working this one out. NH2 Na O N aq. HCl O N N Cl Answer: We’ll use the Ph abbreviation here, because the benzene ring structure does not change during the course of mechanism. The overall _________________________________________________________ Electrophilic Aromatic Substitution 53 Diazonium cation: R N N Diazotization: The reaction in which a primary amine is converted into a diazonium salt. + Nitrosonium cation: N=O structure change requires addition of a new nitrogen atom (probably from NO+) and loss of two hydrogen atoms. Mechanism: Diazotization of a Primary Amine H2O O H N H2O O O H N OH O Protonate O- twice to make leaving group (water) N Ph NH2 N O Ph H OH2 N N O H2O H N N Ph Ph N N H OH2 Ph H H2O H N N Ph OH Ph N N N N OH2 Departure of leaving group gives diazonium cation Protonation converts OH into H2O, a leaving group Deprotonation gives N=N O Protonate oxygen to make water (leaving group) Remove one N-H bond OH N Nitrosonium cation (an electrophile) Water leaves to form nitrosonium cation H Form N-N bond O OH2 Ph N N Diazonium cation resonance contributors Note that water acts as a proton shuttle in this mechanism, serving to both deliver and take away a proton at various times. If you have already studied carbonyl chemistry (topics 33-35) you are probably familiar with the idea of a proton shuttle molecule. The details of your mechanism may not be the same as shown above, the difference being the timing of the various steps. Other diazotization mechanisms are acceptable as long as no fundamental mechanism or structure rules are violated. It is even possible that several similar mechanisms, all giving the same product, are operating in the same reaction vessel simultaneously. Diazo coupling: The EAS reaction of a diazonium cation with another aromatic ring to form a diazo compound. Now we are ready to consider the EAS portion of the butter yellow synthesis. A reaction in which a benzenediazonium cation and another aromatic ring couple to form a diazo compound is called diazo coupling. What is the mechanism? _________________________________________________________ Electrophilic Aromatic Substitution 54 Think Ahead Question 30.28 Write the mechanism for the diazo coupling of phenyldiazonium chloride and N,N-dimethylaniline to give butter yellow. N(CH3)2 N2 Cl N + H2O N N(CH3)2 Answer: The reaction involves replacement of a benzene ring hydrogen by a diazonium cation (an electrophile), so the standard EAS mechanism is likely. N(CH3)2 N N N(CH3)2 N(CH3)2 H Ph N OH2 Diazonium cation adds to form most stable arenium ion N N N Ph Ph Deprotonation restores aromaticity Diazonium cations are weak electrophiles, so the nucleophilic benzene ring component of the diazo coupling reaction must have at least one strong activator (OH, OR, NH2, NR2, etc.). (Review activators in section 30.5B if necessary.) Concept Focus Question 30.26 Give the major product(s) of these diazo coupling reactions. (a) Phenol with benzenediazonium chloride (b) Nitrobenzene with benzenediazonium chloride OCH3 (c) NH2 + NaNO2 aq. HCl NH2 (d) HO NaNO2 + aq. HCl N(CH3)2 _________________________________________________________ Electrophilic Aromatic Substitution 55 30.15 Electrophilic Aromatic Substitution Reactions with Aromatics Other Than Benzene A. Polycyclic Aromatics So far in this topic we have seen numerous examples of EAS reactions on benzene rings. However benzene is not the only aromatic molecule. (Review sections 10.xx and 10.xx if needed.) EAS reactions can involve aromatic systems other than benzene, but what are the mechanisms and products? For example, consider the case of EAS nitration of naphthalene. aq. HNO3 mononitration product(s) aq. H2SO4 Naphthalene Think Ahead Question 30.30 How many mononitronaphthalene isomers are possible when naphthalene is reacted with an aqueous mixture of HNO3 and H2SO4? Which isomer is the major product? Answer: EAS involves replacement of hydrogen atoms, so we need to determine how many different hydrogen atoms there are. Models are a very useful way to do this. Build two models, label the hydrogens in question then see if the models are superposable. Coincident hydrogens are equivalent; non-coincident hydrogens are not equivalent. If they cannot, the hydrogens are not equivalent. (You have already encountered this skill if you studied NMR spectroscopy in topic 14 or 15.) Using this comparison we find that the eight hydrogen atoms of naphthalene can be divided into two sets. H1, H4, H5 and H8 are all equivalent. H2, H3, H5 and H6 are a different equivalent set. H8 H1 H7 H2 H6 H3 H5 Equivalent hydrogens H1, H4, H5 and H8 H2, H3, H6 and H7 H4 Naphthalene has only two different hydrogen types, so EAS nitration can produce only two mononitro isomers. _________________________________________________________ Electrophilic Aromatic Substitution 56 NO2 NO2 aq. HNO3 + aq. H2SO4 1-nitronaphthalene 2-nitronaphthalene Which nitronaphthalene isomer is the major product? Just like all of the previous EAS reactions we have studied so far, the regioselectivity of this reaction is controlled by the stability of the arenium ion intermediates. Start by drawing all the resonance contributors for the arenium ions derived from electrophilic attack at the 1- and 2positions. (Recall from section 30.9 that the nitronium cation, NO2+, is the electrophile responsible for EAS nitration.) Attack at the 1-position: H NO2 H H NO2 NO2 H NO2 H H NO2 NO2 NO2 H NO2 _________________________________________________________ Electrophilic Aromatic Substitution 57 Attack at the 2-position: NO2 H H NO2 NO2 H H H NO2 NO2 NO2 H NO2 Attack at the 1-position yields an arenium ion with seven resonance contributors, four of which retain an aromatic sextet. Attack at the 2position gives an arenium ion that only six resonance contributors, and only two of these retains an aromatic sextet. The carbocation with the greatest number of significant resonance contributors is most stable, so the arenium ion derived from attack at the 1-position is formed more quickly than the arenium ion derived from attack at the 2-position. Therefore we predict that 1nitronaphthalene is the major product. This prediction is verified by experimental results. Concept Focus Question 30.27 Give the major product(s) of the following EAS reactions. O (a) + Cl Ph AlCl3 aq. HNO3 (b) aq. H2SO4 (c) aq. HNO3 aq. H2SO4 NO2 (d) aq. HNO3 aq. H2SO4 _________________________________________________________ Electrophilic Aromatic Substitution 58 B. Heteroaromatics Now lets consider EAS reactions on aromatic molecules whose aromatic ring structure includes atoms other than carbon. Furan, an aromatic molecule with an oxygen atom in the ring, is a good place to start. Think Ahead Question 30.30 Predict the major product when furan is treated with acetyl chloride and aluminum chloride. O O + + O AlCl3 Cl O O O 2-Acetylfuran Furan 3-Acetylfuran Answer: This is another Friedel-Crafts acylation reaction, a typical EAS reaction whose major product is controlled by the stability of the carbocation intermediate. (The reaction does not occur on a benzene ring so the intermediate is not really an arenium ion, but rather a heteroarenium ion.) So we need to determine the resonance contributors for the two heteroarenium ions. Read about the numbering system used to label the positions of the furan ring in section 43.xx. Attack at the 2-position: CH3 H C O H O O O O H Cl O AlCl3 O O O O Attack at the 3-position: O H O Cl3Al Cl O H CH3 C O O O O O _________________________________________________________ Electrophilic Aromatic Substitution 59 Which heteroarenium ion is more stable, and therefore leads to the major product? Electrophilic addition at the 2-position gives a heteroarenium ion with three resonance contributors. The heteroarenium ion derived from addition at the 3-position has only two resonance contributors. In both cases the most significant resonance contributor is the oxonium ion, with its full octet on every atom. Everything else being equal, the cation with the greatest number of significant resonance contributors is most stable, so we predict that attack at the 2-position is favored. Once again, experimental results agree with our prediction. Take a moment to consider what we have just accomplished in this section of this topic. Before this point in our studies, we have paid little attention to naphthalene or furan, yet we successfully predicted the products of their reactions, simply by applying the principles of reactivity and mechanism we have learned previously. This is a hallmark of understanding organic reactions from a conceptual, mechanistic viewpoint: Much of the unknown can be predicted with a fair degree of certainty. Now apply what you have learned to some additional heteroaromatic EAS examples. Concept Focus Question 30.28 Provide the major products of each reaction. I2, HgO (a) O O (b) Cl CH3 AlCl3 O O O (c) O N BF3 H Hint: Acetic anhydride reacts with BF3 to generate an acylium cation, just like acetyl chloride plus AlCl3. aq. HNO3 (d) aq. H2SO4 N N(CH3)2 (e) Cl2 FeCl3 N _________________________________________________________ Electrophilic Aromatic Substitution 60 (f) N CH3 N Br2 FeBr3 30.16 In the Real World: Industrial Synthesis of FD&C Red #40 Look around you and see how colorful your world is. Clothing, food, cosmetics and even the cover of this textbook are alive with various shades of vivid color. Some of this coloration is natural, but most of it (especially in clothing) is added during manufacture. The coloration is provided by dyes, a broad group of molecules (and mixtures) whose structures are as varied as the colors they produce. The first dye was Tyrian purple, isolated by the ancient Greeks and Romans in small quantities from a gastropod mollusk (M u r e x brandaris). About 12,000 snails yielded about 1.5 grams of dye. Because of the labor involved in its production, Tyrian purple was reserved for royalty. Indigo, used to dye denim, is similar in structure to Tyrian purple. Note extensive conjugation for both dye molecules, and the how the addition of two bromine atoms causes a change in color. (Review section 9.xx on the relationship between color and molecular structure.) O H N X X X = Br, Tyrian purple X = H, Indigo N H O The first synthetic dye was made by accident, but it was an accident with exceptional consequences. In 1856, William Perkin was trying to make the anti-malarial drug quinine by oxidation of p-toluidine (cheaply available in large quantities from coal tar, an industrial waste product). However the fundamental concepts of organic structure and complex molecule synthesis were (to be polite) naive at the time, Perkin’s synthesis yielded no quinine. This does not mean his synthesis failed completely, because what he did produce turned out to be the first synthetic red dye, called mauveine (or Perkin’s mauve). This synthetic dye made Perkin a very rich man at a young age, and launched the modern dye industry, and arguably all of industrial-scale organic chemistry. As you examine the structure of the major component of mauveine, think about its conjugation as well as the complexity of the reaction mechanism that produces it. _________________________________________________________ Electrophilic Aromatic Substitution 61 OCH3 OH X N NH2 N Quinine K2Cr2O7 Coal tar aq. H2SO4 H3C N H2N N CH3 CH3 p-toluidine N H Major component of mauveine Modern dyes provide a rainbow of color but are based on just a few fundamental molecular skeletons. The bright colors of these dyes result from structures with extensive conjugation. For example, diazo dyes are usually red, orange or yellow. Adding or changing substituents (typically phenol and amine groups) changes the color. Sulfonic acid groups may also be added to control water solubility and dying properties. A fundamental concept that we have encountered time and time again throughout this topic is that EAS is a useful reaction to replace an aromatic or heteroaromatic hydrogen atom with some other substituent. This transformation is important for small-scale laboratory work, but also of great significance for synthesis on an industrial scale to prepare hundreds of pounds (or hundreds of tons) of some organic material. EAS is especially important to the dye industry, where diazo coupling is a key step in the synthesis of many diazo dyes. Na O3S OH N N Diazo link provides extended conjugation and red color OCH3 CH3 SO3 Na _________________________________________________________ Electrophilic Aromatic Substitution 62 Some food, drug and cosmetic products owe their bright colors to diazo dyes. For example, the vivid red color of maraschino cherries (in canned fruit cocktail) is due to FD&C Red No. 40 (also called Allura Red AC). Lets practice what we have learned about EAS by thinking about the industrial scale synthesis of this substance. Think Ahead Question 30.31 FD&C Red No. 40 could be made by two different diazo coupling reactions. Give the structures of the starting materials for both routes. Briefly justify why both of these diazo coupling reactions will give the desired regioisomer. Answer: Don’t get discouraged by the complexity of the target molecule. An EAS reaction (diazo coupling, in this case) follows the same rules on molecules simple and complex. Start with what you know about diazo coupling. As we saw in section 30.14, the diazo coupling reaction links a diazonium salt (ArN2+) with a nucleophilic aromatic ring by the EAS mechanism. Diazonium cations are weak electrophiles, so the nucleophilic aromatic ring must have one or more strongly activating groups. Both the benzene and naphthalene rings that are attached to the diazo group of FD&C Red No. 40 have strong activators (OH and OCH3), so either (in principle) can be the nucleophile in the EAS reaction. First we consider the case in which the diazonium cation is derived from a naphthalene amine. HO3S Na O3S OH OH N + comes from N NH2 OCH3 OCH3 CH3 CH3 SO3 Na SO3H Why does this reaction give the diazo regioisomer that corresponds to FD&C Red No. 40? Recall that when directing groups compete, the strongest activator dominates (section 30.8). Methoxy is a strong activator, methyl is a weak activator and the sulfonic acid group is a strong deactivator. The methoxy activates the two positions ortho to it, _________________________________________________________ Electrophilic Aromatic Substitution 63 but the position between OCH3 and SO3H is more sterically hindered. (The position para to the OCH3 is blocked). Therefore the diazo coupling should give the desired regioisomer. Methoxy directs here OCH3 X sterically hindered CH3 SO3H In the second diazo coupling possibility, the diazonium salt is derived from 4-amino-5-methoxy-2-methylbenzenesulfonic acid. HO3S Na O3S OH OH + N comes from N NH2 OCH3 OCH3 CH3 CH3 SO3H SO3 Na Why does this reaction give the diazo regioisomer that corresponds to FD&C Red No. 40? Any electrophile would preferentially attack the most electron rich of the two naphthalene rings, especially a weak electrophile such as a diazonium cation. Hydroxy is a strong activator (electron donor) whereas the sulfonic acid group is a deactivator so the naphthalene ring carrying the OH group is attacked. This ring has three possible sites for EAS, so which is preferred? We saw in section 30.15A that electrophilic attack at the 1-position of naphthalene gives an arenium ion that is more stable than attack at the 2-position. In addition, OH is an ortho/para director, which reinforces attack at the desired position. Therefore we have faith that the diazo coupling reaction we have designed will give the desired product as the major one. HO3S Attack on this ring disfavored by SO3H Naphthalene favors attack here OH HO directs attack here _________________________________________________________ Electrophilic Aromatic Substitution 64 We end this topic by asking you to apply your new knowledge of EAS chemistry to work even further backwards in the synthesis of FD&C Red No. 40, designing route to the two molecules used in the diazo coupling from structurally simpler starting materials such as anisole and naphthalene. Summary 30.1 Why Should I Study This? Electrophilic aromatic substitution (EAS) is the general name for a reaction in which a hydrogen atom of an aromatic ring is replaced by some other substituent. The new substituent enters as an electrophile. The reaction is the most important way to add new groups to a benzene ring, and has wide application in organic synthesis. 30.2 Why is Benzene Different: Substitution Versus Addition Electrophilic attack on alkenes and alkynes generally leads to addition products whereas electrophilic attack on benzene gives substitution products (Ar-H → Ar-Elec). 30.3 A General Mechanism for Electrophilic Aromatic Substitution The general EAS mechanism can be divided into three portions. First an electrophile is generated (if necessary). The electrophile adds to the aromatic ring to form a resonance-stabilized carbocation (arenium ion). This is the rate-determining step of the EAS mechanism. The final step is deprotonation of the arenium ion to restore aromaticity. Resonance contributors H B Elec Elec Elec Arenium ion Electrophile attacks benzene ring forming resonance - stabilized arenium ion. Carbocation fate: deprotonation. Only this carbocation fate restores aromaticity. 30.4 Halogenation EAS bromination and chlorination are achieved by reaction with Br2 or Cl2 and a Lewis acid such as FeBr3 or FeCl3. The Lewis acid increases the electrophilicity of Br2 or Cl2, but may not be _________________________________________________________ Electrophilic Aromatic Substitution 65 necessary if the benzene ring has a strong activator such as OH. Iodine can be introduced by reaction with I2 and an oxidant such as HgO or HNO3. There is no EAS reaction for direct fluorination of a benzene ring. Br Cl Br2, FeBr3 Cl2, FeCl3 I2, HgO I 30.5 Substituent Effects: Electron-Donating Groups Electron-donating groups stabilize adjacent carbocations by resonance and inductive effects, so they are ortho/para directors. Because they accelerate an EAS reaction by carbocation stabilization and by enhancing benzene ring nucleophilicity, they are also activators. Ortho/para directors include alkyl groups, and any group that can stabilize a carbocation by resonance with lone pairs or pi bonds. All ortho/para directors are activators, except the halogens, which are ortho/para directing deactivators. Table 30.3 lists common ortho/para directors. X (ortho/para director) X X X Elec Elec + + Elec Elec Ortho major product Para major product Meta minor product 30.6 Substituent Effects: Electron-Withdrawing Groups Electron-withdrawing groups destabilize adjacent carbocations by resonance and inductive effects, so they are meta directors. Because they decelerate an EAS reaction by carbocation destabilization and by decreasing aromatic ring nucleophilicity, they are also deactivators. All electron-withdrawing groups are meta directors. Table 30.4 lists common meta directors. _________________________________________________________ Electrophilic Aromatic Substitution 66 X (meta director) X X X Elec Elec + + Elec Elec Meta major product Ortho minor product Para minor product 30.7 Substituent Effects: The Halogens Halogen substituents are the only deactivating ortho/para directors. 30.8 Substituent Effects: Effect of Multiple Substituents If the aromatic ring undergoing EAS has more than one substituent, the order of influence is: Strong activator (most influential) > weak activator > weak deactivator > strong deactivator (least influential). 30.9 Nitration A nitro group (-NO2) can be introduced by reaction with aqueous HNO3 and aqueous H2SO4. The electrophile is the nitronium cation, NO2+. NO2 aq. HNO3 aq. H2SO4 30.10 Sulfonation A sulfonic acid group (-SO3H) can be introduced by reaction with SO3 in H2SO4. The electrophile is either SO3 or HSO3+. Desulfonation is accomplished by heating with aqueous acid. SO3H SO3, H2SO4 H3O+ 30.11 Friedel-Crafts Alkylation An alkyl group (-R) can be introduced by reaction of the corresponding alkyl chloride (R-Cl) with AlCl3. This is the Friedel-Crafts alkylation reaction. Other carbocation sources such as alkenes and alcohols can also be used. The carbon bearing the chlorine must be sp3. The intermediate is a carbocation or _________________________________________________________ Electrophilic Aromatic Substitution 67 incipient carbocation (RCl-AlCl3 complex). Carbocation rearrangement is possible. The reaction fails if the aromatic ring is strongly deactivated. R RCl AlCl3 30.12 Friedel-Crafts Acylation O CR) can be introduced by reaction with an acid An acyl group ( chloride and AlCl3. This is the Friedel-Crafts acylation reaction. The electrophile is an acylium ion (R-C≡O +). Carbocation rearrangement does not occur. The reaction fails if the aromatic ring is strongly deactivated. O R O R Cl AlCl3 30.13 The Clemmensen and Wolff-Kishner Reductions A ketone can be reduced to a methylene group, or an aldehyde to a methyl group by treatment with Zn/Hg in aqueous HCl (Clemmensen reduction) or with H2NNH2 and KOH (WolffKishner reduction). O R R Zn/Hg, aq. HCl or H2NNH2, KOH 30.14 Diazo Coupling Diazo compounds (Ar-N=N-Ar) can be synthesized by reacting a diazonium cation (ArN2+; the electrophile) with another aromatic ring (ArH). The nucleophilic aromatic ring must be strongly activated. The diazonium cation is produced by diazotization, the reaction of a primary aromatic amine (ArNH2) with the nitronium cation (+N=O), produced from NaNO2 and aqueous acid. Because of their extended conjugation, diazo compounds are colored red, orange or yellow, and some have found use as dyes. _________________________________________________________ Electrophilic Aromatic Substitution 68 NH2 N2 X NaNO2, aq. HCl N N (X = strong activator) X 30.15 Electrophilic Aromatic Substitution Reactions With Aromatics Other Than Benzene Aromatic rings other than benzene can also participate in EAS reactions. The same features that influence EAS of substituted benzenes also influence these cases. 30.16 In The Real World: Synthesis of FD&C Red No. 40 FD&C Red No. 40 is a diazo dye whose structure may appear complex, but its synthesis can be readily dissected by applying the mechanistic concepts and logic learned in this topic. New Terms Electrophilic aromatic substitution (page 1) Arenium ion (page 3) Ipso hydrogen (page 6) Ortho/para director (page 19) Activator (page 24) Deactivator (page 26) Meta director (page 27) Nitronium cation (page 36) Sulfonic acid (page 37) Friedel-Crafts alkylation (page 40) Incipient carbocation (page 42) Acylium ion (page 46) Friedel-Crafts acylation (page 46) Clemmensen reduction (page 50) Wolff-Kishner reduction (page 50) Diazo compound (page 53) Diazonium cation (page 53) Nitrosonium cation (page 53) Diazotization (page 53) Diazo coupling (page 54) _________________________________________________________ Electrophilic Aromatic Substitution 69