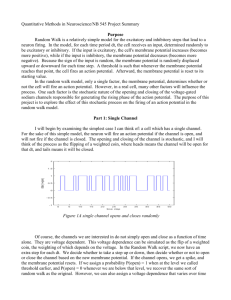
BK potassium channels facilitate high-frequency firing
advertisement

859 J Physiol 580.3 (2007) pp 859–882 BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells Ning Gu, Koen Vervaeke and Johan F. Storm Institute of Basal Medicine, Department of Physiology and Centre of Molecular Biology and Neuroscience, University of Oslo, PB 1103 Blindern, N-0317 Oslo, Norway Neuronal potassium (K+ ) channels are usually regarded as largely inhibitory, i.e. reducing excitability. Here we show that BK-type calcium-activated K+ channels enhance high-frequency firing and cause early spike frequency adaptation in neurons. By combining slice electrophysiology and computational modelling, we investigated functions of BK channels in regulation of high-frequency firing in rat CA1 pyramidal cells. Blockade of BK channels by iberiotoxin (IbTX) selectively reduced the initial discharge frequency in response to strong depolarizing current injections, thus reducing the early spike frequency adaptation. IbTX also blocked the fast afterhyperpolarization (fAHP), slowed spike rise and decay, and elevated the spike threshold. Simulations with a computational model of a CA1 pyramidal cell confirmed that the BK channel-mediated rapid spike repolarization and fAHP limits activation of slower K+ channels (in particular the delayed rectifier potassium current (I DR )) and Na+ channel inactivation, whereas M-, sAHP- or SK-channels seem not to be important for the early facilitating effect. Since the BK current rapidly inactivates, its facilitating effect diminishes during the initial discharge, thus producing early spike frequency adaptation by an unconventional mechanism. This mechanism is highly frequency dependent. Thus, IbTX had virtually no effect at spike frequencies < 40 Hz. Furthermore, extracellular field recordings demonstrated (and model simulations supported) that BK channels contribute importantly to high-frequency burst firing in response to excitatory synaptic input to distal dendrites. These results strongly support the idea that BK channels play an important role for early high-frequency, rapidly adapting firing in hippocampal pyramidal neurons, thus promoting the type of bursting that is characteristic of these cells in vivo, during behaviour. (Received 16 December 2006; accepted after revision 9 February 2007; first published online 15 February 2007) Corresponding author J. F. Storm: Department of Physiology and Centre of Molecular Biology and Neuroscience (CMBN), University of Oslo, PB 1103 Blindern, N-0317 Oslo, Norway. Email: jstorm@medisin.uio.no Potassium (K+ ) channels are by far the most diverse class of ion channels, and are important for regulating neuronal excitability and signalling activity in a variety of ways (Hille, 2001). Since these channels mediate outward K+ currents and increase the membrane conductance, they tend to hyperpolarize the cell and attenuate the effects of excitatory stimuli. Potassium channels are therefore normally regarded as inhibitory, i.e. they reduce neuronal excitability. The clinical relevance of the dampening effect of K+ channels is illustrated by the several familial forms of diseases in humans that are caused by genetic K+ channel defects, including benign neonatal convulsions (Steinlein, 2001) and episodic ataxia (Browne et al. 1994). This paper has online supplementary material. C 2007 The Authors. Journal compilation C 2007 The Physiological Society Accordingly, genetic suppression of K+ channel activity in mice also causes epilepsy (Smart et al. 1998; Peters et al. 2005). Also pharmacological blockade of K+ channels, e.g. with 4-aminopyridine (Rutecki et al. 1987) or barium (Hotson & Prince, 1981), readily causes epileptic seizures. A variety of inhibitory K+ channel effects have been described in rodent CA1 hippocampal pyramidal cells, which currently is one of the most intensely studied neuronal types in the mammalian brain (Storm, 1990; Johnston et al. 2000). For example, A- and D-type K+ channels suppress the initial excitability and cause a delay in the onset of spiking (Gustafsson et al. 1982; Storm, 1988). A-type channels also strongly suppress dendritic excitability and the impact of dendritic excitatory synapses (Hoffman et al. 1997; Ramakers & Storm, 2002). Calcium-activated K+ channels of the sAHP type cause DOI: 10.1113/jphysiol.2006.126367 Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 860 N. Gu and others profound feed-back inhibition of the action potential frequency during repetitive firing (Madison & Nicoll, 1982, 1984; Pedarzani & Storm, 1993). Also Kv7/KCNQ/M-type potassium channels contribute importantly to spike frequency adaptation in these cells (Madison & Nicoll, 1984; Gu et al. 2005; Peters et al. 2005). Calcium-activated K+ channels of the SK type have been reported to have a similar role (Stocker et al. 1999), but this appears not to be the case in CA1 pyramidal cells under normal conditions (Gu et al. 2005). However, SK channels have been shown to be activated by Ca2+ spikes (Gu et al. 2005) or by Ca2+ influx through synaptically activated NMDA channels (Cai et al. 2004; Ngo-Anh et al. 2005). BK channels are Ca2+ - and voltage-activated potassium channels with large conductance (also called BKCa , KCa 1.1, MaxiK, Slo) (Blatz & Magleby, 1987; Marty, 1989; Sah & McLachlan, 1992; Vergara et al. 1998). These channels have been shown to mediate rapid spike repolarization and fast afterhyperpolarization (fAHPs) in many types of neurons (Adams et al. 1982; Storm, 1987a,b; Lancaster & Nicoll, 1987; Schwindt et al. 1988; Womack & Khodakhah, 2002). The BK channels were originally also reported to reduce the frequency of action potentials and to limit epileptiform bursts in rat hippocampal CA1 pyramidal neurons (Lancaster & Nicoll, 1987; Alger & Williamson, 1988). However, during previous experiments more than a decade ago, it was noticed that blockade of the BK channels in rat CA1 pyramidal cells slowed down the initial discharge frequency in response to current injection (J. F. Storm, unpublished observations). At that time, this effect was attributed to suppression of the BK channel-dependent rapid spike repolarization and fAHP (Storm, 1987a,b). These effects would be expected to increase inactivation of the spike-generating transient Na+ current (I NaT ) and activate more of the slower K+ currents, thereby enhancing refractoriness and reducing excitability during the immediate aftermath of the first action potential (Shao et al. 1999a). Thus, BK channels seemed to be able to facilitate high-frequency firing in these neurons. Recently, the idea of a facilitory effect of BK channels was supported by a study of epilepsy in humans. It was discovered that an abnormal increase in the BK channel conductance, caused by a gain-of-function mutation in the BK channel α-subunit, underlies human epilepsy and paroxysmal movement disorder (Du et al. 2005). Similarly, dentate granule neurons from mice lacking the β4 BK channel subunit show a gain-of-function for BK channels that sharpen action potentials, thereby facilitating high-frequency firing and leading to temporal lobe seizures (Brenner et al. 2005). Thus, for the first time, epilepsy was found to be caused by an increase in a potassium current, rather than by a decrease. Furthermore, BK channel-deficient mice have recently been shown to have reduced EEG power (Storm et al. 2006). J Physiol 580.3 In the meantime, other reports have also lent support to the notion that suppression of BK channel activity can indirectly convey a reduction of neuronal excitability. Thus, our group found that mice lacking BK channels showed a reduction of fAHP and firing activity in cerebellar Purkinje neurons (Sausbier et al. 2004). Furthermore, in cultured neurons from the cerebral cortex of mice, bursting activity induced by various procedures was inhibited by the BK channel blocker iberiotoxin (IbTX) (Jin et al. 2000). Previously, certain other K+ channels, in particular the Kv3 channels with their unusually fast activating and deactivating kinetics, have been found to facilitate sustained high-frequency firing, by mediating rapid spike repolarization and deep, fast afterhyperpolarizations (Rudy et al. 1999). Thus, blockade of Kv3 channels in interneurons resulted in a cumulative Na+ channel inactivation, thus slowing down the firing rate (Erisir et al. 1999; Lien & Jonas, 2003). Nevertheless, the effects of BK channel activity on excitability and the underlying mechanisms have previously not been examined in a systematic manner, although BK channels are believed to mediate rapid spike repolarization and fAHPs in many types of neurons (Adams et al. 1982; Storm, 1987a, 1990; Lancaster & Nicoll, 1987; Schwindt et al. 1988; Takahashi, 1990; Gho & Ganetzky, 1992; Sah & McLachlan, 1992; Gola & Crest, 1993; Hu et al. 2001; Womack & Khodakhah, 2002; Sah & Faber, 2002; Zhang et al. 2003; Sausbier et al. 2004). Earlier studies have provided seemingly conflicting results. Thus, in one study, blockade of BK channels by a low dose of tetraethylammonium (TEA) or charybdotoxin (ChTX) was found to reduce the fAHP amplitude and increase the initial discharge frequency during a train of action potentials in hippocampal CA1 pyramidal neurons (Lancaster & Nicoll, 1987). Furthermore, BK channel blockade by TEA or ChTX was found to increase the duration of epileptiform bursts and suppress the associated transient AHP (Alger & Williamson, 1988). Later, it was suggested that when the cell is depolarized, BK channels may contribute to the normal post-burst medium afterhyperpolarization (mAHP) and early spike frequency adaptation (Storm, 1989, 1990; Williamson & Alger, 1990). Increased excitability was reported to result from BK channel blockade also in dissociated Purkinje neurons (Swensen & Bean, 2003), rat dorsal root ganglia neurons (Zhang et al. 2003), and spontaneously firing vestibular nucleus neurons (Nelson et al. 2003). This is in apparent contrast to our own previous results, as well as the reduced Purkinje cell excitability and reduced EEG power found in neurons from BK channel knock-out mice (Sausbier et al. 2004; Storm et al. 2006), and the human epilepsy caused by a BK channel gain-of-function mutation (Du et al. 2005). For these reasons, we have undertaken a systematic examination of the effects of BK channels on the C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 BK channels enhance high-frequency firing and adaptation J Physiol 580.3 excitability and firing pattern of CA1 hippocampal pyramidal cells, as well as the underlying mechanisms. We found that blockade of the BK channels significantly slows down the early high-frequency firing, indicating that BK channels facilitate high-frequency firing in these cells. Furthermore, by combining experiments and computational modelling, we found that these effects are mediated through fast spike repolarization, fAHP generation and modulation of other K+ channels and Na+ channel inactivation. This provides an example of how a fast calcium-activated potassium channel type can enhance neuronal excitability through interaction with other ion channels. Some of the data have already been published in abstract form (Gu et al. 2006; Vervaeke et al. 2006a). Methods Slice electrophysiology Slices for sharp-electrode intracellular recording and field potential recording. Young adult male Wistar rats (4–7 weeks of age) were deeply anaesthetized with Suprane before decapitation. Transverse hippocampal slices (400 μm) were cut with a Vibroslicer (Campden Instruments, UK) and maintained in an interface chamber filled with artificial cerebral spinal fluid (aCSF) containing (mm): 125 NaCl, 25 NaHCO3 , 1.25 KCl, 1.25 KH2 PO4 , 1.5 MgCl2 , 1.0 CaCl2 , 16 glucose and saturated with 95% O2 –5% CO2 . The aCSF was heated to 30◦ C during recording, and the temperature was kept constant within ±0.5◦ C. The experimental procedures were approved by the responsible veterinarian of the Institute, in accordance with the statute regulating animal experimentation given by the Norwegian Ministry of Agriculture 1996. Sharp-electrode intracellular recording. Slices were transferred to a recording chamber maintained at 30 ± 0.5◦ C, where they were submerged and perfused with aCSF of the composition described above, except that the concentration of CaCl2 was 1.6 mm and KCl was 3.5 mm. Sharp electrodes were filled with 4.0 mm potassium acetate (resistance 80–140 M, pH 7.35). Intracellular recordings were obtained from CA1 pyramidal neuron somata with the ‘blind’ method. Currentclamp recordings were performed using an Axoclamp 2A amplifier (Molecular Devices Corporation, USA) in bridge mode. Only cells with a stable resting membrane potential more negative than −60 mV and stable action potential amplitudes (> 80 mV) were included. Extracellular field potential recording. Extracellular field potentials were recorded submerged in a chamber maintained at 31 ± 0.5◦ C, with low resistance glass micropipettes filled with aCSF (as described above, 861 except that the CaCl2 concentration was 2.0 mm) and coupled to an Axoclamp 2B amplifier (Molecular Devices Corporation). A tungsten stimulation electrode was placed in the middle of the stratum radiatum while the recording electrode was placed in the pyramidal layer, ∼200–400 μm apart. The stimuli, consisting of a brief current pulse (100 μs duration, repeated every 30 s) elicited two to four population spikes. dl-APV (50 μm) and bicuculline (5 μm) were routinely added to the extracellular solution. The extracellular recording solution was the same as described above except that the CaCl2 concentration was 2.0 mm and the K+ concentration was 4.0 mm. Data acquisition, storage and analysis The data were acquired using pCLAMP 7.0 or 9.0 (Molecular Devices Corporation) at a sampling rate of 5–20 kHz, analysed and plotted with pCLAMP 9.0 and Origin 7.5 (Microcal). Values are expressed as mean ± s.e.m. Two-tailed Student’s t test was used for statistical analysis (P = 0.05). The P values are given in the figure legends. The interspike interval (ISI) was the time measured between the peaks of two consecutive action potentials (APs) within a spike train. The 1st ISI is the time between the peaks of the 1st and 2nd APs, and so on. The instantaneous firing frequency of an ISI is f = 1/ISI (Hz). The cell input resistance was calculated by dividing the steady-state voltage response by the current pulse amplitude (Rinput = V /I). Chemicals and drugs The BK-channel blocker iberiotoxin (IbTX) was obtained from Dr Hans-Guenther Knaus, Innsbruck Medical University, Austria. The M-channel blocker XE991 was obtained from DuPont pharmaceutical company and NeuroSearch, Denmark. DNQX (6,7dinitroquinoxaline-2,3-dione) and TEA (tetraethylammonium) were purchased from Tocris Cookson Ltd (UK). Apamin was purchased from Latoxan (France). The remaining chemicals, including forskolin, were purchased from Sigma-Aldrich Norway AS (Oslo). Substances were bath-applied by adding them to the perfusion medium. Computer simulations Computer simulations were performed using the Surf-hippo neuron simulator, version 3.5a (Graham, 2004). Surf-Hippo is written in Lisp and was run on a Linux workstation. For integrating the circuit equations, Surf-Hippo uses a variant of the Crank Nicholson method as described by Hines (1984). The variable time-step method was used to speed up the simulations C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 862 N. Gu and others (Borg-Graham, 2000), with a maximum voltage error criterion of 0.05 mV. We used a computer model that is derived from the Borg-Graham CA1 pyramidal neuron model (Borg-Graham, 1999), which has been refined through collaborative studies by using our experimental data on discharge patterns, AHPs, subthreshold resonance and oscillations (Shao et al. 1999a; Hu et al. 2002; Gu et al. 2005; Vervaeke et al. 2006b). This model reproduces quite accurately the spiking behaviour of these neurons (cf. Shao et al. 1999a) as well as the AHPs following single action potentials or spike trains (Gu et al. 2005; Vervaeke et al. 2006b). We refer to the full description of the model in the supplementary material. Briefly, the cell was represented by five compartments, comprising an isopotential soma (diameter 20 μm) and a dendritic cable (total length 800 μm and diameter 5 μm) consisting of four segments of equal length. Dendritic spines were not explicitly modelled, but their contribution to the linear dendritic load was modelled by raising the dendritic segment capacitance and reducing the dendritic membrane resistance (Rall et al. 1992). The soma and the dendritic segments had a membrane resistance of 20 and 6 k cm2 , respectively, while the capacitance was 1.0 and 1.5 μF cm−2 . A uniform intracellular resistance of 100 cm was assumed (Stuart & Spruston, 1998). With these values, the model was able to account for the input resistance and whole-cell capacitance measured in our experiments. The active conductances included persistent and transient Na+ currents (I NaP , I NaT ), four voltage-gated potassium currents (I A , I D , I DR , I M ), a fast inactivating Ca2+ - and voltage-dependent K+ current (I BK ) (Shao et al. 1999a), two voltage-gated calcium currents (I CaN and I CaL ), a hyperpolarization-activated non-specific cation current (I h ) and a slow Ca2+ -activated potassium current: the sAHP current (I sAHP ). The soma was divided into three subcellular compartments to model the differential and localized intracellular Ca2+ dynamics. The outer membrane shell consists of two juxta-membrane shells to reproduce the Ca2+ channel dependence of the BK- and sAHP current, in particular a co-localization of the channels underlying I CaN and I BK (Borg-Graham, 1987). The rest of the somatic volume is occupied by a core compartment. As Ca2+ ions enter through the calcium channels they diffuse among the subcellular compartments and undergo instantaneous buffering while a Ca2+ pump causes efflux of Ca2+ ions into the extracellular space. [Ca2+ ] in all compartments was initially set to 50 nm. Calculation of all currents was based on the extended Hodgkin–Huxley formalism with rate constants derived from a single barrier thermodynamic gating particle model (Borg-Graham, 1987). Exceptions are I NaT and I BK , which were calculated according to a more generalized Markov scheme (Shao et al. 1999a; Borg-Graham, 1999; J Physiol 580.3 Vervaeke et al. 2006b). The simulation temperature was 30◦ C. In Fig. 10, the accumulative I BK inactivation was removed by increasing the transition rate from the inactivated state (I) to the closed state (C). This was done by shifting the V 1/2 of the squeezed exponential describing this transition from −120 mV to −50 mV (eqn (1)). ⎛ −1 ⎞−1 V − V1/2 ⎠ αI→C (V ) = ⎝τ0 + α0 + exp k (1) In Fig. 10D, the recovery of inactivation of I NaT was varied by changing the V 1/2 of the squeezed exponential describing the transition from the inactivated (I) to the closed state 1 (C1) to −53, −55, −57 and −59 mV for the panels from left to right, respectively. The excitatory synapse was calculated as I syn = g syn (E rev – V m ), with reversal potential (E rev ) of 0 mV and the unitary synaptic conductance (g syn , peak conductance 0.3 μS) was described by a difference of exponentials with time constant of 0.1 ms for the rising phase and 5.0 ms for the decay phase. This synapse was inserted 300 μm from the soma. Results Intracellular recordings Somatic intracellular recordings with sharp microelectrodes were obtained from 53 CA1 pyramidal neurons, in stratum pyramidale of rat hippocampal slices from young adult male Wistar rats (4–7 weeks of age). All cells were recorded in current-clamp mode, in normal aCSF (with [K+ ]o = 3.5 mm and [Ca2+ ]o = 1.6 mm) at 30.0 ± 0.5◦ C. The average resting membrane potential (V rest ) and input resistance (Rinput ) were −68.2 ± 1.6 mV and 40.7 ± 2.1 M, respectively. To standardize our tests, the membrane potential prior to stimulation of the cell was normally maintained at a constant level (−60 mV) by injection of DC current (Figs 1–5, 7 and 9), but tests at −70 mV were also performed. TEA reduced the early high-frequency firing, but did not affect low-frequency firing Several potassium channels that are important for spike repolarization and spike frequency control in mammalian central neurons can be reversibly blocked by TEA. This is true for several voltage-gated K+ channels such as Kv3 (Rudy & McBain, 2001), some Kv7 channels (KCNQ or M-channels) (Hadley et al. 2000) and the Ca2+ -activated, voltage-dependent BK channels (Storm, 1987a; Lancaster & Nicoll, 1987). TEA, being a small, water-soluble molecule, has the advantage that it readily diffuses in and out of the slice, C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 J Physiol 580.3 BK channels enhance high-frequency firing and adaptation producing rapid effects and recovery upon washout. In our experiments, bath-application of a low dose of TEA (100 μm) produced clear, reversible effects on spike repolarization and frequency in all the CA1 pyramidal cells tested (Fig. 1). From a membrane potential of −60 mV, we injected two depolarizing current pulses: one brief and strong (50 ms, 0.8 nA), the other longer but weaker (100 ms, 0.2 nA), in order to evoke high- and low-frequency firing, respectively (Fig. 1Aa). A 6 s interval between the two pulses ensured that the slow afterhyperpolarization (sAHP) following the first pulse fully decayed before the onset of the second. At the start of each experiment, the current intensities were adjusted to evoke five spikes within 50 ms (strong pulse), followed by four spikes within 100 ms (weak pulse), and the intensities then remained constant for the rest of the experiment. Interestingly, application of 100 μm TEA always reduced the initial discharge frequency in response to the strong pulse – an effect that may seem unexpected for a K+ channel blocker (Fig. 1A, left, insets). Thus, the initial spike 863 frequencies were significantly reduced by TEA: both the instantaneous frequency for the first interspike interval (ISI) (Fig. 1B and C), and the average frequency of the 1st to the 3rd ISIs (Fig. 1D and E). Thereby, the 2nd to 4th action potentials were delayed, often preventing the 5th action potential from occurring within the 50 ms pulse (Fig. 1Ab, left). The effects of TEA were fully reversible (Fig. 1B and D). This demonstrated that the K+ channel blocker TEA actually reduced the excitability of these neurons, in response to strong stimulation. In contrast, the response to the weaker pulse was not noticeably changed by 100 μm TEA (Fig. 1Ab, right; Fig. 1B and D, open symbols; Fig. 1C and E, right). These results seem to indicate that TEA-sensitive potassium channels can enhance the excitability and increase the firing rate of CA1 pyramidal neurons. However, this effect is dependent on the discharge frequency, as it was only seen during high-frequency spike trains. What is the mechanism underlying the enhanced excitability? TEA (100 μm) did not significantly change Aa 1st - 3rd ISI Control st 1st - 3rd ISI nd 1 AP 2 AP -60mV TEA b -60mV Washout c 20 mV 20 ms -60mV High freq. Low freq. TEA 100uM C 200 (1st ISI) frequency (Hz) 300 100 0 0 10 20 Time (min) 30 (1st ISI) B frequency (Hz) 0.5 nA 250 200 150 100 50 0 High freq. * Low freq. NS Ctrl TEA Ctrl TEA TEA 100uM (1st - 3rd ISI) 150 High freq. Low freq. 100 50 0 0 10 20 Time (min) 30 200 150 (1st - 3rd ISI) 200 Averaged freq. (Hz) E D Averaged freq. (Hz) Figure 1. Tetraethylammonium (TEA) reduced high-frequency discharge Sharp intracellular electrode recordings from rat CA1 pyramidal cells. Aa, repetitive firing at two different frequencies was evoked by somatic injection of depolarizing current pulses. Left: a strong (0.8 nA) 50 ms long pulse evoked 5 action potentials (APs). Right: a weaker (0.2 nA) 100 ms long pulse evoked 4 APs. The current pulses, which were separated by a 6 s interval, were repeated at constant intensities throughout the experiment, while the background membrane potential was kept at −60 mV by injecting steady depolarizing current. Ab, bath application of TEA (100 μM) clearly reduced the high-frequency firing rate and spike number (left: the number of APs was reduced from 5 to 4), but had no significant effect on low firing frequency (right). Ac, washout of TEA for 10 min reversed the effect. B, time course of the 1st ISI firing frequency. (•: high frequency; e: low frequency). C, summary data showing that 100 μM TEA significantly reduced the 1st ISI high firing frequency (∗ : n = 7; washout: n = 4; P < 0.05), but had no effect on low-frequency firing (NS: n = 7; washout: n = 4; P > 0.05). D, time course of the averaged firing frequency of the 1st–3rd interspike intervals (ISIs): •, high frequency; e, low frequency. E, summary data showing that 100 μM TEA significantly reduced the mean firing frequency of 1st–3rd ISI in response to the strong (0.8 nA, 50 ms) pulse (∗∗ : n = 7; washout: n = 4; P < 0.01), but had no effect on the low firing frequency in response to the weak (0.2 nA, 100 ms) pulse (NS: n = 7; washout: n = 4; P > 0.05). High freq. ** Low freq. NS 100 50 0 Ctrl TEA Ctrl TEA C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 864 N. Gu and others the holding current (i.e. the steady current required to hold the cell at −60 mV), in any of the cells tested (n = 7). Nor did TEA significantly change input resistance of the cells, as measured by injecting negative current pulses (n = 4). However, TEA (100 μm) produced a significant spike broadening and blockade of the fast afterhyperpolarization (fAHP) in all cells tested. (P < 0.01, n = 7), in accordance with previous results (Storm, 1987a,b; Shao et al. 1999a). Figure 2 shows the effect of IbTX on the early firing during a 50 ms spike train, evoked by a strong depolarizing current pulse (0.8–1.2 nA, 50 ms). Again, the current intensity was adjusted to trigger five spikes during each pulse under control conditions, but thereafter the intensity was kept constant. Application of IbTX (100 nm) slowed the repolarization of the first two action potentials and increased the duration of the 1st ISI (Fig. 2Aa–c), thus reducing the instantaneous firing frequency (Fig. 2C). However, IbTX did not significantly change the cell input resistance, nor the DC current required to hold the background potential at −60 mV (n = 7, P > 0.05, data not shown). Similar results were obtained in all cells tested (Fig. 2E, ∗ P < 0.05, n = 6). Figure 2D shows that the frequency of the initial discharge (1st to 3rd ISIs) was reduced by BK channel blockade, thus reducing the number of spikes during the 50 ms pulse from 5 to 4 (see Fig. 2Bb). Similar effects were observed in all cells tested The BK channel blocker iberiotoxin (IbTX) reduced high-frequency firing Since TEA blocks several K+ channel types, more selective blockers are needed to identify the channel types mediating its effects. To test whether BK channels, which are sensitive to TEA in the micromolar range (Storm, 1987a), were involved, we used the highly selective BK channel blocker iberiotoxin (IbTX) (Galvez et al. 1990). Aa Control AP1 J Physiol 580.3 c IbTX b AP1 AP2 AP2 Overlay AP2 AP1 20 mV 2 ms fAHP fAHP Ba c b 20 mV 20 ms 0.5 nA D IbTX 100nM 200 150 100 0 5 10 15 20 Time (min) 25 200 200 frequency (Hz) 250 E Averaged 1st-3rd ISI frequency (Hz) 1st ISI frequency (Hz) C IbTX 100nM 150 100 50 0 5 10 15 Time (min) 20 25 control IbTX * ** 150 100 50 0 1st ISI 1st-3rd ISI Figure 2. The BK channel blocker IbTX reduced early high-frequency firing A and B, typical sharp intracellular electrode recordings from a rat CA1 pyramidal cell. Ba, a high-frequency train of 5 action potentials (APs) was evoked by a 0.9 nA, 50 ms long depolarizing current pulse, starting from a membrane potential of −60 mV (maintained by steady depolarizing current injection). Bb, 13 min after application of iberiotoxin (IbTX, 100 nM), the cell fired only 4 APs in response to an identical current pulse. A shows the expanded traces of the 1st and 2nd APs taken from B, showing that the 2nd was significantly delayed after application of IbTX (Ac). Blockade of BK channel broadened the action potentials and eliminated the fast afterhyperpolarizations (fAHPs; arrow). Summary time course showing that 100 nM IbTX reduced the instantaneous firing frequency of the 1st ISI from 169 ± 5 Hz to 146 ± 7 Hz (C, n = 6), and the averaged 1st−3rd ISI frequency from 122 ± 4 Hz to 103 ± 3 Hz (D, n = 6). E, summary data from all cells tested (n = 6): comparison of the 1st ISI frequencies and averaged 1st−3rd ISI frequencies before and after application of IbTX (n = 6, each. ∗ P = 0.016, ∗∗ P = 0.001). C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 BK channels enhance high-frequency firing and adaptation J Physiol 580.3 (Fig. 2E, ∗∗ P < 0.01, n = 6). (Wash-out of IbTX was not attempted, since previous experience indicates that this cannot normally be obtained in slices; Shao et al. 1999a.) These experiments are in agreement with previous findings (J. F. Storm, unpublished data), supporting the hypothesis that blockade of BK channels, rather than increasing the excitability of the CA1 hippocampal neurons, tends to reduce their initial firing rate during high-frequency discharge. frequency greater than 150 Hz. However, the experiments with TEA, suggest that blockade of BK channels does not affect the firing rate at lower frequencies (Fig. 1). Nevertheless, the apparent lack of effect in that case might be due to a balance between opposing effects of different currents blocked by TEA, e.g. M-current (Hadley et al. 2000) and BK current. To test these hypotheses, we used a similar protocol as in Fig. 1A (right), injecting weak pulses (0.2–0.4 nA) that evoked four action potentials within 100 ms (Fig. 3B). With this procedure, application of IbTX (100 nm) did not produce any significant change in the 1st ISI; there was neither a prolongation nor a shortening of the interval (Fig. 3Aa–c), in contrast to the effect of IbTX on the high-frequency response (Fig. 2Aa–c). Nevertheless, IbTX clearly slowed the repolarization of the first two action potentials (Fig. 3Ac), and also decreased Blockade of BK channels failed to affect the early firing at low frequency The above results suggest that BK channel activity promotes high-frequency firing, at least at average frequencies greater than 100 Hz and, for the 1st ISI, at Aa Control IbTX b AP2 AP1 865 AP1 Overlay c AP1 AP2 AP2 20 mV 5 ms Ba b c 20 mV 20 ms −60 mV 0.5 nA E D IbTX 100nM 100 50 0 0 5 10 15 20 25 150 150 frequency (Hz) 150 Averaged 1st-3rd ISI frequency (Hz) 1st ISI frequency (Hz) C IbTX 100nM 100 50 0 100 Time (min) 5 10 15 20 Time (min) 25 NS 50 0 0 control IbTX NS 1st ISI 1st-3rd ISI Figure 3. Application of IbTX had little or no effect on low-frequency firing A and B, typical sharp intracellular electrode recordings from a rat CA1 pyramidal cell. Ba, the cell fired a train of 4 APs in response to a 0.4 nA, 100 ms long depolarizing current pulse, with the preceding membrane potential held at −60 mV by steady-state current. Aa–c show expanded traces taken from 1st to 2nd action potentials of Ba–c. Ab and Bb were recorded 14 min after bath application of 100 nM IbTX. Bc, overlaid traces showing that IbTX did not significantly change the firing rate of the cell. The current pulse was constant (0.4 nA). Ac, IbTX caused spike broadening of the two first spikes, but the interspike interval (ISI) was not changed. C, summary time course from all the cells tested shows that IbTX did not significantly change the instantaneous firing frequency of the 1st ISI: (before IbTX: 68.1 ± 8.5 Hz; after application of IbTX: 65.2 ± 8.5 Hz; n = 4). D, time course of averaged 1st–3rd ISI frequency (41.3 ± 4.1 and 40.8 ± 3.4 Hz, before and after application of IbTX, respectively) (n = 4). E, summary graph showing no significant IbTX effect on either the 1st ISI or the averaged 1st–3rd ISI frequency (NS: P > 0.05). C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 866 N. Gu and others the spike frequencies in response to strong depolarizing pulses in these experiments as shown in Fig. 2, confirming that the toxin blocked BK channels. Similar results were obtained in all cells tested in these ways (Fig. 3E, n = 6 for the 1st ISI and the 1st to 3rd ISIs, NS; P > 0.05). Figure 3C and D summarizes the average time courses for all the cells tested in this way. All the experiments presented in Figs 1–3 were performed while holding the cell at a slightly depolarized potential: −60 mV. However, it is possible that this depolarization might increase the resting inactivation of the Na+ channels, leading to a larger effect of BK blockade than at the normal resting potential of these cells, which is close to −70 mV. To test for this possibility, we repeated such experiments at a background potential of −70 mV. We found that the effects of IbTX at −70 mV were essentially the same as at −60 mV. Thus, also at −70 mV, IbTX application significantly reduced the initial high-frequency firing frequency in response to strong current injection (n = 6, P < 0.01; for 1st ISI, the frequency was reduced from 203 ± 14 to 152 ± 19 Hz; for the 1st to 3rd ISI, the average frequency was reduced from 144 ± 10 to 109 ± 10 Hz). Also, at −70 mV, IbTX failed to affect the low-frequency firing in response to weak current injection (average frequency of 1st to 3rd ISI: 79.3 ± 7.5 Hz before IbTX; 77.4 ± 6.3 Hz after IbTX application; P > 0.05, n = 4). The experiments and model simulations in this study were mainly performed at temperatures close to 30◦ C (range: 29–31◦ C), as is common for hippocampal slice recordings. To test whether the observed effects also occur at physiological temperatures, we also performed a few experiments at ∼37.5◦ C. At this recording temperature, the maximal early spike rate was higher (up to ∼300 Hz), but IbTX still significantly reduced the early discharge rate by more than 20%. Thus, in the two cells tested, the average initial firing frequency for 1st to 3rd ISIs was reduced, with convincing time courses, from 194 to 153 Hz (by 22%) and from 263 to 208 Hz (by 21%), respectively (data not shown). These results indicate that the effect of BK channels on neuronal excitability is frequency dependent. However, why did the BK channels only facilitate the high-frequency firing while having no detectable effect on the low-frequency firing? To investigate this, we took a closer look at the first spikes during high-frequency discharge. Effects of IbTX on action potential slopes during early high-frequency firing Figure 4Aa shows a typical example of the first two action potentials of a high-frequency spike train (same procedure as in Fig. 2; 1st ISI, 5.7 ms). In normal medium, there was always a clear fast AHP (arrow) after the first spike. Application of IbTX eliminated this fAHP, and the 1st J Physiol 580.3 ISI was increased from 5.7 to 7.2 ms (the frequency was reduced from 175 to 139 Hz; Fig. 4Ab). In parallel, the decay of the first two spikes were slowed by IbTX (Fig. 4Ac), due to loss of repolarizing BK current (Storm, 1987a; Shao et al. 1999a). Similar results were obtained in all five cells tested (summarized in Fig. 4Da and b, and Fa and b). We then switched our attention to the rising slope of the action potentials, reasoning that the IbTX-induced spike broadening and loss of the fAHP might affect sodium channel inactivation. Since the upstroke of the spikes is due to the charging of the cell membrane capacitance by the voltage-dependent transient Na+ current, the rising slope of the spike reflects the size of the net Na+ current during the fast depolarization. Figure 4Ba and b shows superimposed traces of the two first spikes; the insets show the rising slopes at an expanded time scale. While the rising slopes of the first spike were not affected by IbTX (insets of Fig. 4Ba, summary time course in Ca, and summary data in Cb), the rising slope of the 2nd spike was significantly reduced by IbTX (see inset of Fig. 4Bb, summary time course in Ea). Such an effect was observed during high-frequency spike trains in all five cells tested in this way (Fig. 4Eb, P < 0.05). In contrast, IbTX had no detectable effect on the rising slope of 1st and 2nd action potentials during low-frequency firing, i.e. four spikes in 100 ms, as in Fig. 3 (n = 5; P > 0.05 for both 1st and 2nd spike, data not shown). Furthermore, during high-frequency firing, IbTX shifted the threshold of the 2nd action potential to a more depolarized potential (on average 3.6 mV shift; n = 5, P < 0.05), while the threshold of the 1st spike remained unchanged (n = 5, P > 0.05). These results indicate that blockade of BK channels reduced the rate-of-rise and elevated the threshold of the second action potential in the train, presumably because broadening of the first spike combined with fAHP blockade caused increased Na+ channel inactivation and reduced recovery before the second spike. In contrast, IbTX did not affect the upstroke of the first spike itself, indicating that the toxin did not directly affect the sodium channels. f–I curves: IbTX reduced the spike frequency gain in the high-frequency range To determine the BK channel contribution to spike frequency regulation at a range of different firing frequencies, a series of depolarizing current pulses of different intensities were injected into each cell before and after IbTX application. The resulting discharge frequencies (f ) versus injected current (I) were plotted as f–I curves (Fig. 5A and B). At the lowest frequencies (< 25 Hz), IbTX had no detectable effect on the f–I plots (Fig. 5A and B; •, control; e, IbTX). At higher frequencies, however, the f–I curves obtained after IbTX application showed a clear decrease in slope compared with the curves C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 BK channels enhance high-frequency firing and adaptation J Physiol 580.3 Modelling f–I curves: BK channels facilitate high-frequency firing obtained in the same cells in normal medium, before IbTX was applied. This was seen both for the f–I curves representing the instantaneous frequency of the first ISI in the train (Fig. 5A) and for those representing the averaged frequencies of the first three interspike intervals (Fig. 5B). Thus, for both types of plots, the effect of IbTX was stronger at higher intensities of injected current, i.e. the effect of BK channel blockade increased with the overall spike frequency (Fig. 5A and B). These results confirm that BK channels enhance the excitability of CA1 pyramidal cells by promoting high-frequency initial discharge, in a manner that is itself frequency dependent. Ca Control fAHP b Da 7.2 ms IbTX 20 mV c b AP1 rise slope (%) AP2 AP1 5.7 ms 1 ms Ea AP1 AP2 rise slope (%) Overlay Ba To explore the mechanisms underlying the BK channel-induced increase in firing frequency, we performed numerical simulations using a computer model of a rat CA1 pyramidal neuron derived from a previous model (Borg-Graham, 1999). Our model is described in detail in Methods (Computer simulations) and further justification of the parameter values is given in Vervaeke et al. (2006b). Briefly, the model contained the following active conductances: persistent and transient Na+ currents (I NaP , I NaT ), four voltage-gated AP1 decay slope (%) Aa 867 AP2 b IbTX IbTX 20 mV 1 ms ▲ AP2 decay slope (%) Fa 200 200 IbTX 100nM 150 100 100 50 50 0 0 5 200 150 10 15 Time (min) 20 25 b 0 200 100 50 50 0 5 200 150 10 15 Time (min) 20 25 200 100 50 50 5 200 10 15 Time (min) 20 25 b 50 10 15 Time (min) IbTX 20 ** 150 50 5 * CTRL 100 0 IbTX 0 100 0 CTRL 200 IbTX 100nM 150 ** 150 IbTX 100nM 0 IbTX 0 b 100 0 CTRL 150 IbTX 100nM 100 0 NS 150 25 0 CTRL Figure 4. Effects of IbTX on the 1st and 2nd action potential slope during early high-frequency firing Aa–c, recordings of the first two action potentials during a high frequency spike train, elicited by a brief, depolarizing current step (0.8 nA, 50 ms). Aa, in normal medium, the ISI between the 1st and 2nd spikes was 5.7 ms (firing rate: 175 Hz). Note the fAHP following the 1st AP (arrow). Ab, application of the BK channel blocker IbTX (100 nM) delayed the onset of the 2nd action potential (ISI: 7.2 ms; frequency: 139 Hz). Ac, overlaid traces of Aa and Ab, comparing the 1st and 2nd APs evoked by identical current pulses (0.8 nA) before and after application of IbTX. Ba, the decay slope of the 1st AP was reduced after application of IbTX (dashed line), whereas the rising slope remained unchanged (see inset at expanded time scale, X: 0.2 ms; Y: 50 mV). Bb, both the rising slope (see inset) and the repolarization slope of the 2nd AP were slowed down by IbTX (dashed line), and there was a slight elevation of the spike threshold (). Ca–Fa, normalized average time courses (%) from all the cells tested: IbTX effects on the rising slope (Ca) and decay slope (Da) of the 1st AP, and the rising slope (Ea) and decay slope (Fa) of the 2nd AP. Cb–Fb, summary graph from all the cells tested, showing that BK channel blockade significantly reduced rising slope of the 2nd AP· Eb (∗ : n = 5, P < 0.05), as well as the decay slopes of both 1st AP (Db,∗∗ : n = 5, P < 0.01) and 2nd AP (Fb, ∗∗ : n = 5, P < 0.01). In contrast, IbTX had no significant effect on the rising slope of the 1st AP (Cb, NS: n = 5, P > 0.05). C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 IbTX 868 N. Gu and others potassium currents (I A , I D , I DR , I M ), a fast inactivating Ca2+ - and voltage-dependent K+ current (I BK ), two voltage-gated calcium currents (I CaN and I CaL ), a hyperpolarization-activated non-specific cation current (I h ) and a slow Ca2+ -activated potassium current (I sAHP ). Also, the soma was divided into three subcellular compartments to model localized intracellular Ca2+ dynamics. By varying the BK channel density, we explored the effects on the input–output properties of these cells by simulating f–I curves (G BK : 0, 10, 20, 40, 80, 130, 200, 400, 600 and 800 pS μm−2 ). Depolarizing current pulse injections of increasing amplitude were simulated at the soma (pulse duration: 1 s). Before each current pulse, the J Physiol 580.3 membrane potential was set to −60 mV by steady current. Increasing the BK channel density clearly promoted higher firing frequencies, both for the 1st ISI (Fig. 5C) and for the average frequency of the first three intervals (Fig. 5D). Reduction of the firing frequency by blocking BK channels is not dependent on activation of M-, sAHP- or SK-channels Several mechanisms might contribute to the reduced firing frequency after blocking BK channels. In particular, the broadened action potential probably causes more Ca2+ influx to occur, in particular since Ca2+ influx predominantly happens during the spike repolarization B 200 175 175 Average frequency 1st -3rd ISI (Hz) 200 150 125 100 75 50 NS 25 0 C * 0.1 0.2 ** 0.3 0.4 0.5 Frequency 1st ISI (Hz) 250 225 200 175 150 125 100 75 50 25 0 GBK = 800 pS/ m GBK = 130 pS/ m 2 2 GBK = 0 pS/ m 2 0.05 0.10 0.15 0.20 0.25 0.30 0.35 I (nA) * NS 125 100 75 50 25 D I (nA) control IbTX 150 0 0.6 0.1 0.2 0.3 0.4 0.5 0.6 I (nA) Average frequency 1st - 3rd ISI (Hz) Frequency 1st ISI (Hz) A 250 225 200 175 150 125 100 75 50 25 0 GBK = 800 pS/ m GBK = 130 pS/ m 2 2 GBK = 0 pS/ m 2 0.05 0.10 0.15 0.20 0.25 0.30 0.35 I (nA) Figure 5. BK channel effects on spike frequencies: results from IbTX experiments and computer simulations A, action potential frequency of the first interspike interval (1st ISI) in response to a series of 1.0 s long depolarizing current pulses (0.1–0.6 nA, in steps of 0.05 nA) in normal medium (•) and after adding 100 nM IbTX ( e). The plot shows the mean firing frequencies from all CA1 hippocampal pyramidal cells tested (n = 7). IbTX clearly reduced the high-frequency firing in response to strong current pulses (≥ 0.2 nA; ∗ P < 0.05; ∗∗ P < 0.01), but had no significant effect on the low firing rate in response to 0.15 nA pulses (NS: P > 0.05). B, similar to A, but showing the average frequencies of the first three intervals (1st–3rd ISI). IbTX significantly reduced the spike frequencies in response to current pulses > 0.35 nA (∗ : n = 7, P < 0.05), but had no significant effect at the lower current intensities (0.15–0.3 nA; NS: n = 7, P > 0.05). C and D, computer simulations with a CA1 pyramidal cell model showed that BK channels promote high-frequency firing. The f–I relations of the 1st ISI (C) and of the average of the first 3 interspike intervals (D, 1st–3rd ISI) are plotted for different BK channel conductance densities (GBK : 0, 10, 20, 40, 80, 130, 200, 400, 600 and 800 pS μm−2 ). Current pulses of increasing amplitude were applied at the soma (pulse duration: 1 s). The membrane potential prior to each current pulse was held at −60 mV by steady current injection. The continuous lines represent the standard model with IBK present (130 pS μm−2 ) or blocked (0 pS μm−2 ). C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 BK channels enhance high-frequency firing and adaptation J Physiol 580.3 phase. Therefore, blockade of BK channels would be expected to increase the Ca2+ -activated K+ current which underlies the slow afterhyperpolarization (I sAHP ), which would reduce the firing frequency. We used the model to predict the effects of interactions between I BK and I sAHP on the f–I curves (Fig. 6A and B). Blocking I sAHP only affected the lower frequency range of the f–I curve because the sAHP conductance is rather small. Thus, at higher frequencies this conductance is negligible compared with the total conductance during the interspike trajectory. Our simulations with and without I sAHP showed that the BK channels induced a virtually identical increase of firing frequency in the upper frequency range, whether I sAHP was present or not. Thus, effects mediated by I sAHP could not account for the observed effects of BK channel blockade by IbTX (Fig. 5A and B). Alternatively, the spike broadening could activate more M-current (I M , mediated by KCNQ/Kv7 channels) which underlies the medium afterhyperpolarization (mAHP) in these cells and is known to strongly dampen excitability (Storm, 1989; Peters et al. 2005; Gu et al. 2005). Therefore, we also explored the role of I M on the input–output A 225 Control no IBK 200 no IsAHP 175 no IsAHP + no IBK 150 125 100 75 50 25 0 225 Control no IBK 200 no IsAHP 175 no IsAHP + no IBK 250 Average Frequency ISI-1-3 (Hz) Frequency ISI-1 (Hz) properties by simulating f–I curves. Blocking I M in the model strongly increased the firing frequency, in particular in the lower frequency range (Fig. 6C and D), as reported previously (Madison & Nicoll, 1984; Storm, 1989; Gu et al. 2005; Peters et al. 2005). Nevertheless, the inclusion of BK channels still induced virtually identical increase in firing frequencies in the upper frequency range whether I M was present or not. In order to test experimentally whether I M and I sAHP were important for the BK channel effect on firing frequency, we first blocked I M and I sAHP and then performed the same type of experiment as shown in Figs 1, 2 and 4. Thus, we evoked a high-frequency spike train by injecting a current pulse, and blocked BK channels by adding 100 nm IbTX. (In three experiments, I M and I sAHP were blocked by 10 μm XE991 and 50 μm forskolin, respectively; in one experiment, I M and I sAHP were both blocked by 20 μm carbachol; all drugs were bath-applied.) In all the four cells tested in this way, IbTX caused a pronounced reduction in the initial spike frequency, and the effect had a convincing time course (Fig. 7Da). For the 1st and 2nd ISI, the effect was essentially the same as B 250 150 125 100 75 50 25 0 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.05 0.10 0.15 0.20 0.25 0.30 0.35 I (nA) 225 Control no IBK 200 no IM 175 no IM + no IBK 150 125 100 75 50 25 0 225 Control no IBK 200 no IM 175 no IM + no IBK 250 Average Frequency ISI-1-3 (Hz) 250 Frequency ISI-1 (Hz) I (nA) D C 869 150 125 100 75 50 25 0 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.05 0.10 0.15 0.20 0.25 0.30 0.35 I (nA) I (nA) Figure 6. Computer simulations: reduction in firing frequency by BK channel blockade is not dependent on activation of I M or I sAHP A and B, f–I relations of the first interspike interval (A, ISI-1) and of the average of the first 3 interspike intervals (B, ISI-1–3) from simulations with IsAHP present (dashed lines) or blocked (continuous lines). The f–I relations were simulated as described in Fig. 5. C and D, similar to A and B, but here the f–I relations were plotted from simulations with IM present (dashed lines) or blocked (continuous lines). C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 870 N. Gu and others seen without blockade of I M and I sAHP (Figs 1, 2 and 4), thus confirming the prediction of the model: that I M and I sAHP are not important for the ability of BK channels to facilitate high-frequency firing. However, in the cells where I M and I sAHP had been blocked, the IbTX-induced spike broadening and the depolarization during the interspike intervals were even more severe (Fig. 7A and B) than with I M and I sAHP intact (Figs 1, 2 and 4), probably because I M , and even I sAHP , may contribute to repolarization of late, broadened spikes and during ISIs. As the spikes late in the train became severely blunted and broadened, it was difficult to interpret the effects on the late firing frequency. Finally, we also tested the possibility that Ca2+ -activated + K channels of the SK type underlie the reduction in firing frequency in response to BK channel blockade. In six cells the SK channels were blocked by 100 nm apamin after (n = 4) or before (n = 2) adding 100 nm IbTX. In the cells that were first treated with IbTX, apamin had no detectable effect on the spike frequency, indicating that SK channels did not contribute appreciably to spike frequency regulation in spite of the IbTX-induced spike broadening (n = 4, data not shown). Conversely, in the cells first treated with apamin, the IbTX-induced reduction in spike frequency was undiminished, thus confirming that SK channels did not mediate this effect (n = 2, data not shown). These results confirm and extend our previous finding that SK channels do not contribute appreciably to the mAHP or spike frequency regulation in CA1 pyramidal cells (Gu et al. 2005). Control IbTX 1st ISI A J Physiol 580.3 20mV 2 ms -60 mV 0 5 10 15 Time (min) b 250 200 IbTX 100nM 150 100 50 0 5 10 15 Time (min) b IbTX 100nM 250 200 150 100 50 0 0 5 10 15 Time (min) 20 100 ** 50 Control IbTX 250 200 ** 150 100 50 0 20 300 Control IbTX 150 0 20 2nd AP rise slope (mV/ms) 2nd AP rise slope (mV/ms) 1st ISI frequency (Hz) 1st AP decay slope (- mV/ms) 50 0 Da IbTX 100nM 100 0 Ca b 150 1st ISI frequency (Hz) 1st AP decay slope (- mV/ms) Ba Control IbTX 300 250 200 150 100 50 0 * Figure 7. Blockade of BK channel reduced the burst firing frequency in the presence of XE991 and forskolin A, sample traces from a sharp-electrode current-clamp recording showing the first three action potentials (1st–3rd spikes) evoked by a 0.8 nA current pulse, before and after bath-application of 100 nA IbTX. The perfusion media contained XE991 and forskolin, to block Kv7/KCNQ/M- and sAHP-currents, respectively. Ba–Da, time courses of the effects of IbTX (100 nA, same experiment as in A) on the decay slope of the 1st spike (Ba), the rise slope of the 2nd spike (Ca), and the instantaneous firing frequency of 1st ISI (Da). Similar results were observed in all the cells tested with IbTX (100 nA, n = 3), as shown by the column graphs in Bb, Cb and Db (∗ P < 0.05, ∗∗ P < 0.01). C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 J Physiol 580.3 BK channels enhance high-frequency firing and adaptation 871 Analysing the roles of ionic currents during repetitive firing: modelling with different BK channel densities I (nA) 12 8 4 0 I (nA) 0 −3 −6 −9 −12 (–) 40 20 0 −20 −40 −60 1.00 0.75 0.50 0.25 0.00 I (nA) Vm (mV) A 2 1 0 GBK: 600 pS μm−2 GBK: 130 pS μm−2 No IBK voltage 20 mV 5 ms IBK INa INa (I-state) IA I (nA) 2 I (nA) 4 0.8 0.6 0.4 0.2 0.0 I (nA) 0.04 0.02 0.00 B IDR 0 dV/dt (mV/ms) Next, we used the model to study the roles of various ionic currents during repetitive firing (Fig. 8). A short spike train evoked by injection of a 50 ms long depolarizing current pulse at the soma (mean spike frequency ∼200 Hz) was simulated with three different BK channel densities: (1) with a BK channel density that probably is close to normal (130 pS μm−2 ; continuous black lines in Fig. 8) and was used in our standard model (Fig. 6); (2) with an increased BK channel density (600 pS μm−2 : dotted lines); and (3) without any BK current (red lines). Figure 8 shows the BK current (I BK ; previously dubbed I CT , by Storm, 1990 and Shao et al. 1999a), the sodium current (I Na ), the inactivation state of I Na (I-state), the A-current (I A ), the delayed rectifier potassium current (I DR ), the M-current (I M ), and the slow AHP current (I sAHP ). I D is not plotted in Fig. 8 because it did not contribute appreciably to spike repolarization under the conditions simulated here. It is a not a large current and is largely inactivated at potentials positive to ∼−65 mV. Therefore, the remaining I D was too small to contribute appreciably. At the presumed normal BK channel density (130 pS μm−2 ; black lines), the first spike in the train is mainly repolarized by I BK , I DR and I A , but later in the spike train, I BK and I A inactivate while I DR plays a larger role (Shao et al. 1999a). When I BK was blocked (red lines), the spike was broadened, causing the other voltage-gated K+ currents (I DR , I A and I M ) to become more strongly activated. Nevertheless, the first spike was considerably broadened and the fAHP suppressed. Together, these effects caused a stronger inactivation of I Na and, in particular, a far less complete recovery from inactivation of I Na between the spikes (see I Na , I-state). This, in turn, caused a slight attenuation of the peak I Na during the second to fourth spikes (I Na trace). Therefore, the peak amplitude and rise slope of these spikes were also reduced (phase plot, Fig. 8B), in agreement with our experimental results. Thus, the amplitude of the 2nd AP was reduced from 69.4 ± 2.9 to 63.9 ± 3.5 mV (n = 6, P < 0.01), whereas the 1st AP amplitude remained unchanged (82.0 ± 2.5 and 81.3 ± 2.9 mV, before and after IbTX, respectively, n = 6, P > 0.05, data not shown) (see also Fig. 4Ac and Bb). The incomplete recovery from inactivation of I Na raised the spike threshold slightly (arrows in Fig. 8B), which contributed to prolonging the interspike intervals, i.e. reducing the firing rate. However, the strongest effect on firing rate was mediated via the delayed rectifier K+ current, I DR . Note that I DR is by far the largest current during the interspike intervals at higher frequencies. I DR represents a current whose molecular identity is not exactly known, although Kv2 subunits are probably involved (Du et al. 2000), and a complete kinetic description is lacking. Therefore, in our previous IM IsAHP 2nd Spike 450 Threshold 300 150 0 −150 −60 −40 −20 0 Vm (mV) 20 40 Figure 8. Computer simulations of a spike train and the response of various ionic currents, while varying the BK channel density A, somatic responses to a current pulse (50 ms, 0.4 nA). The membrane potential prior to the pulse was held at −60 mV by steady current. The simulations were repeated for 3 different BK channel densities: 0 (red), 130 (black), and 600 pS μm−2 (dashed black). The first four spikes in the train are shown. Note that the delayed rectifier potassium current (IDR ) increases considerably during the interspike intervals when the spikes are broadened as a consequence of a reduced BK channel density (0 pS μm−2 , red). Also, INa does not recover completely from inactivation when the spikes get broader (INa , I-state). B, phase plot of the second spike. Note the increased threshold and reduced rise slope and spike peak when IBK is blocked (red arrow). C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 872 N. Gu and others (Vervaeke et al. 2006b) and present model, I DR is based on a combination of voltage-clamp (e.g. Du et al. 2000) and current-clamp data (e.g. Storm, 1987a; Shao et al. 1999a; see also Vervaeke et al. 2006b). Our model predicts that the spike broadening caused by blocking I BK strongly increases I DR , thus increasing refractoriness after the spike and decreasing the firing frequency in the upper frequency range. The spike broadening also caused an increased activation of I M and I sAHP, but these currents are too small (compared with I BK , I Na and I DR ) to have any substantial effects on the initial high-frequency firing rate (Fig. 8A, lower two rows). When the I BK density was increased to 600 pS μm−2 (dotted lines in Fig. 8), the spikes became narrower and, hence, I DR was substantially reduced, not only the I DR peak during the spike, but also the I DR tail current between the spikes. In parallel, I Na inactivation was reduced, and recovery from inactivation more rapid. Together, these mechanisms produced a briefer refractory period and, hence, higher discharge frequencies. Thus, our simulations provide an explanation for how BK channels facilitate high-frequency firing: the BK channels do this mainly by ensuring rapid spike repolarization and narrow spikes, which prevent strong activation of I DR and promote rapid recovery from I Na inactivation after each spike. The BK current generates early spike frequency adaptation Several lines of evidence indicate that the BK current in CA1 pyramidal cells is transient, i.e. it decays within less than 20 ms during sustained depolarization (Zbicz & Weight, 1985; Storm, 1990; Shao et al. 1999a,b; Halvorsrud et al. 1999; Gu et al. 2006; Vervaeke et al. 2006a). Therefore the current was dubbed ‘I CT ’, i.e. transient Ca2+ -activated K-type current (Storm, 1990). This inactivation contributes to frequency-dependent spike broadening and the rapid decay of the fAHP during a high-frequency discharge (Shao et al. 1999a). We reasoned that the inactivation of the BK current will also necessarily influence spike frequency and, in particular, spike frequency adaptation. As outlined above, the spike frequency is reduced when I BK inactivates, due to enhanced I DR activation and Na+ channel inactivation causing enhanced refractoriness. Therefore, the inactivation of I BK during the initial spikes in a highfrequency train should cause spike frequency adaptation. In order to test this hypothesis, we performed slice experiments (Fig. 9A–C) as well as computer simulations (Fig. 9D–F). To standardize the conditions, in both experiments and simulations, the cells were held at −60 mV and current steps were injected to keep the instantaneous spike frequency of the first interval (1st ISI) at about 100 Hz (Fig. 9Aa and Da). J Physiol 580.3 In the slice experiments, BK channel blockade by bath application of 100 nm IbTX, caused both a slowing of the initial discharge frequency, as we saw before (Figs 2 and 4), and also a reduction in the spike frequency adaptation. In order to compare adaptation in spike trains with the same initial frequency, a current step with stronger intensity was injected into the cell, giving a frequency of ∼100 Hz for the 1st ISI also after addition of IbTX (dashed lines in Fig. 9A). In this situation, the frequency of the next spikes (2nd and later ISIs) was clearly increased after adding IbTX compared with the control condition, indicating that BK channel blockade strongly reduced the early spike frequency adaptation (Fig. 9Ab and Db). Similar results were obtained in all the six CA1 pyramidal cells tested. The data from six experiments are summarized in Fig. 9B and C (∗∗ P < 0.01; NS, P > 0.05). Similar results were also obtained while holding the cells at −70 mV (n = 6, see above; data not shown). In the computer simulations we used a similar protocol and got similar results (Fig. 9D–F). Thus, the early spike frequency adaptation was strongly reduced when I BK was omitted from the model (Fig. 9Db) compared with when I BK was included (Fig. 9Da). This is in close qualitative agreement with the experimental results from IbTX application (Fig. 9A–C), although there are minor quantitative differences (at least compared with the example shown in Fig. 9A), e.g. in the current needed to obtain ∼100 Hz for the 1st ISI, the spike amplitude, late spike frequencies, and the exact shape of the ISI and frequency plots (Fig. 9E and F). No attempt was made to tune the model in order to reduce these differences; the simulation results shown in Fig. 9D–F are the ones that were first obtained when the model was used for this purpose. Thus, the modelling results represent a prediction of the experimental results shown in Fig. 9A–C, rather than just a post hoc explanation of them. Our prediction was also confirmed by earlier experiments and modelling. Thus, in Fig. 8A, the third ISI is virtually identical with BK current (black, continuous line: 130 pS μm−2 ) and without (red line), whereas the first ISI is about 30% longer when the BK current is missing, indicating reduced adaptation. A similar effect was seen experimentally in response to IbTX (Fig. 2B). Figure 8A also shows that the activation of I DR increased strongly following the 2nd spike compared with the 1st spike with normal BK current, whereas there was much less increase in I DR in the simulation without BK channels (red line). In addition, Fig. 8A also shows that the inactivation of I Na (I Na I-state) increased following the 3rd spike compared with the 1st spike with normal BK current (black line), whereas there is no such increase without BK channels (red line; actually the I-state here was slightly reduced). Thus, it seems that because the BK current facilitates spiking but inactivates rapidly, it contributes to early spike frequency adaptation. C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 BK channels enhance high-frequency firing and adaptation J Physiol 580.3 BK current inactivation underlies early spike frequency adaptation Figure 10A outlines the normal, inactivating I BK model, with its closed (C), open (O), and inactivated (I) states. With such an inactivating I BK in the model, injection of a current pulse evoked a high-frequency spike train with prominent spike frequency adaptation (Fig. 10Ca, black upper trace). Here, the fAHP amplitude rapidly decayed, because of rapid inactivation of I BK (Fig. 10Ca, black middle trace). In contrast, in the non-inactivating I BK model, the same current pulse produced a train of spikes that showed much weaker spike frequency adaptation (red plots in Fig. 10Ca and Cb), and each spike was followed by a prominent fAHP (Fig. 10Ca, red upper trace; arrows). In order to test more ‘directly’ our idea that rapid inactivation of the BK current is essential for the contribution of this current to early spike frequency adaptation, we selectively removed the inactivation in the model. By comparing a model with the normal, inactivating I BK and a model in which the inactivation mechanism of I BK was removed (Shao et al. 1999a), we could address this issue in a direct way that was not experimentally feasible (Fig. 10). Experiment Aa Computer model Da 1st - 3rd ISI 873 1st - 3rd ISI Control Control 20 mV 10 ms -60mV 20 mV -60mV 10 ms 1 nA 1 nA 1st - 3rd ISI 1st - 3rd ISI b b IbTX No IBK 20 mV 20 mV 10 ms 10 ms -60mV -60mV 1nA C E 120 40 ** 100 ** 30 20 NS 10 0 80 Control IbTX NS 60 2 ISI number 3 Control No IBK 40 30 ** ** 40 20 10 20 0 1 F 2 ISI number 3 100 80 60 40 20 0 0 1 Control No IBK 120 Frequency (Hz) Control IbTX ISI (ms) 50 Frequency (Hz) ISI (ms) B 1 nA 1 2 3 ISI number 4 1 2 3 ISI number Figure 9. Experiments and computer modelling showed reduced spike-frequency adaptation after BK channel blockade Aa, sample trace showing an early part of a spike train in response to a brief current injection. Before the pulse, the cell was held at −60 mV by DC current injection. Note that in normal medium (Control), the cell showed strong spike-frequency adaptation. Ab, after bath-application of 100 nM IbTX, a current step with stronger intensity was injected to the cell in order to achieve the same firing frequency of the 1st ISI as before IbTX (vertical dashed lines). Note that the 2nd and 3rd interspike intervals (ISIs) were clearly reduced compared with the control condition. B and C, summary plots showing reduced spike-frequency adaptation after BK channel blockade by 100 nM IbTX: B, the 2nd and 3rd ISIs were clearly reduced by IbTX when the 1st ISI was kept constant (NS: n = 6, P > 0.05; ∗∗ : n = 6, P < 0.01). C, spike frequencies of the 2nd and 3rd ISIs were clearly increased by IbTX when the frequency of the 1st ISI was kept constant (NS: n = 6, P > 0.05; ∗∗ : n = 6, P < 0.01). D–F, results from computer simulations of the experiments shown in panels A–C, respectively. C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 4 874 N. Gu and others In this case, I BK showed no attenuation during the spike train (Fig. 10Ca, red middle trace). This result indicates that rapid inactivation of the BK current is essential for its contribution to early spike frequency adaptation. By giving a series of current steps of different intensities, we also plotted the f–I curves (Fig. 10B). Removal of I BK inactivation had essentially no effect on the 1st ISI (Fig. 10B, left panel; note that the red and black plots overlap). In contrast, there was a clear increase of the average frequency of the first four spikes at high stimulus current intensities (Fig. 10B, right panel). This C O I Ca illustrates, again, that removing I BK inactivation did not only increase excitability but also reduced the spike frequency adaptation. Next, we explored how I BK inactivation can affect neuronal excitability during a burst of spikes in response to excitatory synaptic input. Such spike bursts are typical responses of CA1 pyramidal neurons (see e.g. Ranck, 1973; Harris et al. 2001). A synaptic conductance was inserted at the apical dendrite of the model, 300 μm from the soma, and the maximal conductance was adjusted to evoke a burst of spikes at the soma (Fig. 10D). Non inactivating IBK 200 160 120 80 Control IBK 40 0 0.0 ISI-1-3 Frequency (Hz) IBK model 1st ISI Frequency (Hz) B A J Physiol 580.3 Non inactivating IBK 200 160 120 40 0 0.0 0.2 0.4 0.6 Current (nA) 0.2 0.4 0.6 Current (nA) IBK Frequency (Hz) b Vm 60 50 40 30 20 10 Control IBK Non inactivating IBK 1 D Control IBK 80 2 3 ISI no. 4 Control IBK Non–inactivating IBK slower recovery of INaT inactivation 20mV 10ms 20mV 10ms Figure 10. Removing the inactivation of I BK in the model reduced the adaptation and increased the excitability A, diagram of the IBK Markov model states; C, closed state; O, open state; I, inactivated state. B, the rate of the I→C transition was increased sufficiently to fully prevent cumulative IBK inactivation, thus producing a ‘non-inactivating’ BK current (red plot) (see Methods). Left panel: f–I relation of the first interspike interval was not changed by removing IBK inactivation. Right panel: f–I relation for the first 4 spikes (average frequency) was increased by removing IBK inactivation. Ca, top trace: spike train in response to a depolarizing current pulse for IBK with (black) and without (red) inactivation (scale bar, 30 mV). Note the fast afterhyperpolarization (fAHP, arrows) following every spike when inactivation was removed. Middle trace: IBK during the spike train (scale bars: 5 ms and 3 nA). Cb, plot of adaptation based on the simulations shown in Ca. D, in the model, a synapse on the apical dendrite (left cartoon) was activated strongly so that the EPSP evoked a burst of spikes at the soma. Top panels: a series of somatic voltage responses of the model; the recovery rate of INa inactivation was reduced stepwise from fast (left panels) to slow (right panels; see Methods). Bottom panels: same as above, but without inactivation of IBK . C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 J Physiol 580.3 BK channels enhance high-frequency firing and adaptation During such high-frequency bursts, recovery from I NaT inactivation is also expected to be an important factor. To explore the role also of this factor, we also varied the latter parameter. Figure 10D shows four cases, with different rates of recovery from I NaT inactivation (left, fast; right, slow recovery). For each case we determined the effect of removing I BK inactivation. With the fastest I NaT inactivation recovery (Fig. 10D, left panel) removing I BK inactivation reduced the spike frequency adaptation, and increased the number of spikes in the burst. When the I NaT inactivation recovery was slower, the removal of I BK inactivation had stronger effects (Fig. 10D, middle traces). However, when the recovery of I NaT inactivation was made very slow (Fig. 10D, right traces), the I BK effect became less significant. Thus, BK channel inactivation can contribute strongly to the early spike frequency adaptation that is characteristic for the bursting of hippocampal pyramidal neurons in vivo (Ranck, 1973; Harris et al. 2001). 875 logical conditions. To test this, we stimulated the Schaffer collateral fibres by positioning a tungsten stimulation electrode in the middle of stratum radiatum, and recorded the somatic population spike response with an extracellular recording pipette placed in the CA1 pyramidal layer (Fig. 11A). The experiments were performed in the presence of GABAA - and NMDAreceptor blockers and in slightly elevated [K+ ]o (4.0 mm), in order to obtain a purely excitatory synaptic response with multiple population spikes, which remained stable because NMDA-receptor-dependent forms of synaptic plasticity were blocked. The stimulation intensity was increased until a single stimulus evoked an excitatory postsynaptic potential (EPSP) that triggered three to five population spikes, with an interval of ∼5 ms (∼200 Hz) between the first two population spikes under control conditions (Fig. 11B). This kind of response resembles the natural response pattern of CA1 pyramidal neurons, which typically fire two to six spikes at 150–200 Hz during a burst event in rats during behaviour (Ranck, 1973; Harris et al. 2001). Furthermore, an advantage of extracellular field potential recording is that it, being non-invasive, does not disturb the intracellular milieu of the cell. Thus, it avoids artifacts that may afflict intracellular recordings, such as the artificial leak conductance caused by sharp BK channel blockade reduced the firing frequency of synaptically evoked population spikes in stratum pyramidale Finally, we wished to test whether BK channels also increased the firing frequency under more physio- A B Extracellular Recording Stimulation Control IbTx PS3 PS2 0.5 mV PS1 PS3 20 D E PS2 10 PS1 5 0 ISI1 150 100 50 0 10 20 30 Time (min) ISI2 0 Control IbTx NS F 5 5 5 4 4 4 3 3 2 2 2 1 1 1 0 * 0 PS1 3 0 PS2 * PS3 G 1.25 1.00 PS3 PS2 PS1 0.75 0.50 0.25 0.00 10 20 30 Time (min) Decay Slope (mV/ms) 0 Frequency (Hz) PS time (ms) 200 15 2 ms Normalized Decay slope (mV/ms) C Rise slope (mV/ms) Figure 11. Blocking BK channels with IbTX reduced the firing frequency of population spikes in striatum pyramidale A, a tungsten electrode was positioned in the middle of stratum radiatum to stimulate the Schaffer collaterals (stimulation duration, 150 μs; stimulation interval, 30 s). A field recording electrode was positioned in stratum pyramidale. The perfusion medium contained 4.0 mM K+ , bicucculine (15 μM) and APV (100 μM). B, under these conditions, a single stimulus (arrow) evoked 3 population spikes (PS). The stimulation artifact is indicated by an arrow. The amplitude of the stimulus current was adjusted so the interval between the first two population spikes was ∼5 ms (i.e. ∼200 Hz). Bath application of 100 nM IbTX reduced the frequency of the population spikes. C, the average (n = 4) time course of the IbTX effect on the PS latencies following the stimulation artifact (t = 0). D, average time course of the IbTX effect on the frequency of the first and second PS interval (ISI1 and ISI2). E, average time course of the normalized change in decay slope of the three population spikes. F and G, summary data of the IbTX effect on the rise slopes and decay slopes of the three population spikes (n = 4, NS: P = not significant, ∗ 0.01 < P < 0.05, ∗∗ P < 0.01). 0 10 20 30 Time (min) Control IbTx ** 3 3 3 * 2 2 2 1 1 1 0 0 0 PS1 * PS2 C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 PS3 876 N. Gu and others microelectrode impalement (as used in this study), or washout effects of whole-cell recording. Adding IbTX (100 nm) broadened the population spikes and reduced the frequency of the burst (Fig. 11B, dashed and dotted line). The time course of the effects of IbTX on the population spike latencies from the stimulus (spike timing) is plotted, for all slices tested, in Fig. 11C, together with the population spike interval frequency (Fig. 11D), and the decay slope of the population spikes (Fig. 11E). Interestingly, IbTX decreased the decay slope of the first three population spikes but the effect was clearly less on the third than on the first spike (Fig. 11E). This indicates that BK-channel-dependent spike repolarization is most prominent for the first few spikes in a burst, thus causing frequency-dependent spike broadening, in agreement with previous results from our lab (Shao et al. 1999a), as well as with Figs 2–4. Thus, given that CA1 pyramidal cells in the behaving animal mostly fire short, high-frequency bursts of spikes rather than long spike trains, BK channel kinetics seem to be well adapted to allow the type of bursting that is characteristic of these cells in vivo. Discussion Main results By combining slice electrophysiology and computational modelling, we have investigated the functional roles of BK channels in regulation of high-frequency firing in rat CA1 hippocampal pyramidal neurons. We found that BK channels facilitate high-frequency discharge (> ∼100 Hz), thus increasing the spike frequency and spike number in response to strong depolarizing current. By causing rapid spike repolarization and a fast afterhyperpolarization (fAHP), the BK channels mediate their facilitating effect. Thus, the short spike duration caused by I BK limits the activation of other, slower K+ channels (in particular, the delayed rectifier, I DR ) and limits the inactivation of Na+ channels. In addition, the BK-dependent fAHP may promote faster recovery from Na+ inactivation and faster closure of slower K+ channels (I DR ). In contrast to DR channels, regulation of M-, sAHP- or SK-channel recruitment does not seem to contribute appreciably to the facilitating effect of BK channels. Since the BK current itself inactivates rapidly, its facilitating effect diminishes during the initial part of a spike train, thus producing a novel form of early spike frequency adaptation. Furthermore, the function of BK channels is highly frequency dependent. Thus, little or no effect of BK channel blockade was observed at spike frequencies below 40 Hz. In addition to the intracellular recordings of repetitive firing in response to somatic current injection, extracellular field recordings demonstrated that BK channel activity is also important for high-frequency J Physiol 580.3 burst firing in response to excitatory synaptic input to the distal dendrites (Fig. 11). The latter conclusion was also supported by model simulations. Taken together, our results strongly support the idea that BK channels play an important role in facilitating early high-frequency firing and fast adaptation in hippocampal pyramidal neurons, thus promoting the type of bursting that is characteristic of these cells in vivo. Mechanisms whereby BK channels promote high-frequency discharge and bursting BK channels are known to play a major role in spike repolarization and generation of the fAHP in hippocampal pyramidal neurons (Storm, 1987a,b; Lancaster & Nicoll, 1987; Shao et al. 1999a; Hu et al. 2001) as well as in a variety of other neurons (Adams et al. 1982; Schwindt et al. 1988; Sah & McLachlan, 1992; Vergara et al. 1998; Womack & Khodakhah, 2002; Sah & Faber, 2002). Converging evidence indicates that the ability of BK channels to open significantly during a single action potential, i.e. within about 1 ms or less, depends critically on both their close co-localization (within 20–40 nm; Marrion & Tavalin, 1998; Berkefeld et al. 2006) with voltage-gated Ca2+ channels, and on the rapid intrinsic voltage-dependent gating of the BK channels (Storm, 1987b; Lancaster & Nicoll, 1987; Blatz & Magleby, 1987; Marty, 1989; Gola & Crest, 1993; Marrion & Tavalin, 1998; Vergara et al. 1998; Sah & Faber, 2002; Berkefeld et al. 2006). The fast spike repolarization caused by the joint action of BK channels and purely voltage-gated K+ channels, has two main effects: (1) it limits the inactivation of the transient, spike-generating Na+ current (I NaT ), whose degree of inactivation is important for determining the spike threshold and therefore the timing of the next spike; and (2) it limits the activation of slower voltage-dependent K+ conductances that are also activated by the spike. Because of their slow deactivation, the latter are particularly important for determining the interspike intervals. As the M-, SK- and sAHP-currents are slow, important for spike frequency regulation (Madison & Nicoll, 1984; Storm, 1989; Storm, 1990; Pedarzani & Storm, 1993; Gu et al. 2005; Peters et al. 2005), and would be more strongly activated by broadened spikes caused by BK channel blockade, they might be suspected to contribute importantly to the slowing of the discharge caused by BK channel blockers. However, when we tested this idea experimentally, by blocking M-, SK- and sAHP-channels, none of these slow K+ conductances seemed to be important for the BK-dependent facilitation of high-frequency discharge. Our computer model yielded the same result: although both I sAHP and I M had substantial effects on sustained low-frequency firing (in agreement with Madison & Nicoll, 1984; Pedarzani & Storm, 1993; Gu et al. 2005; Peters et al. 2005), their effects on the initial high-frequency firing C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 J Physiol 580.3 BK channels enhance high-frequency firing and adaptation was small, and they contributed essentially nothing to the BK-dependent facilitation (Fig. 6). Instead, our simulations predict that the delayed rectifier, I DR , is the main K+ current contributing to the BK-dependent facilitation of high-frequency firing during brief spike trains or bursts. The main reasons for the dominant role of I DR are evident from Fig. 8: the amount of I DR that is activated by a spike (3–4 nA) is far larger than I sAHP and I M (0.003–0.6 nA), and the tail current of I DR after each spike is sufficiently slow and large to strongly delay the subsequent spike. The A-current, I A , is about as large as I DR during the first spike in the train (∼3 nA), but because of the rapid deactivation kinetics of I A , it decays almost to zero well before the next spike, and has therefore little influence on spike timing or frequency in this situation. These model predictions are consistent with all our experimental data so far, including the lack of effect of SK channel blockade. (Recall that SK channels were not included in this model because they do not seem to contribute measurably to normal repetitive firing (Gu et al. 2005) and we lack sufficiently detailed data for CA1 pyramidal cells.) However, the precise role of I DR must await further experimental testing. In contrast, the role of Na+ inactivation is supported by convergent evidence. Thus, the elevation of the spike threshold and reduced dV /dt of the spike upstroke (following BK channel blockade) are both consistent with this mechanism. In addition to the consequences of BK-dependent rapid spike repolarization discussed above, BK channels also influence the initial discharge frequency by generating the fAHP. On one hand, the fAHP should contribute to refractoriness by hyperpolarizing the cell away from the spike threshold immediately after the spike. On the other hand, the fAHP is expected to promote both rapid recovery from Na+ inactivation and rapid closure of voltage-dependent K+ conductances (in particular I DR ) between the spikes. The relative importance of the two latter factors depends sensitively on the exact voltage- and time-dependences of these two processes, and must await more detailed analysis to be quantitatively determined. However, our experimental results as well as our model simulations indicate that the facilitating effects of BK channels dominate over fAHP-mediated refractoriness, at least for the spike frequency range explored here. Thus, the combined effects of spike duration-induced facilitation (in particular, via reductions in I DR tail current and Na+ inactivation) and fAHP-induced facilitation (via speeding of I DR closure and recovery from Na+ inactivation) are evidently larger than the relatively modest hyperpolarizing effect of the fAHP away from threshold. The explanation outlined above is in accordance with all our electrophysiological and modelling results. In particular, it explains why application of a BK channel blocker strongly reduced the early firing rate (Figs 1 and 877 3). In contrast, BK channel blockade had no detectable effect on discharge frequency or the number of spikes in response to a weak depolarizing current, yielding spike frequencies less than 50 Hz (Figs 1 and 2). Our modelling indicated that the lack of BK channel effects at low discharge frequencies was caused by two factors: (1) a nearly complete recovery from Na+ channel inactivation between the spikes – even after IbTX-broadened spikes – when the frequency is so low; and (2) a minimal effect of the increased activation of DR channels by the broadened spikes, because the DR channels have time to close between the spikes at such low frequencies. Which properties enable BK channels to facilitate high-frequency firing? Which properties make a K+ channel type primarily inhibitory or excitatory/facilitating? Presumably, this is not only determined by the properties of the K+ channel alone, but by the entire biophysical context in which it operates: the properties and distribution of various ion channels within the cell, as well as its passive membrane properties and geometry. Thus, K+ channels of the same type may play different roles in different neuron types, in different parts of the same neuron, and in different situations. Nevertheless, it seems that some general properties are needed to enable K+ channels to facilitate high-frequency firing, and that these rules must also apply to BK channels in CA1 pyramidal cells: (1) the channels should have sufficiently rapid activation kinetics to turn on during a spike; (2) they should produce a large outward current contributing substantially to spike repolarization and/or fast AHP (thus preventing strong Ca2+ influx, strong activation of slower K+ currents, and strong Na+ channel inactivation); (3) they should have deactivation kinetics that are neither too fast nor too slow: i.e. the channels should stay open long enough to ensure rapid, full spike repolarization and a deep, fast AHP (thus promoting rapid voltage-dependent closure of the slower K+ currents and rapid recovery from Na+ channel inactivation); but on the other hand the BK channels should close before the earliest time at which the next spike may occur, in order to avoid delaying it. These requirements place quite strict boundaries on optimal BK channel kinetics. Since an action potential in CA1 pyramidal cells lasts ∼1–2 ms, the BK channels should activate substantially in ∼1 ms and since these cells can fire at rates up to ∼250 Hz, i.e. with ∼4 ms intervals, and the spike lasts ∼1 ms, the BK channels should deactivate nearly completely in < 3 ms, but not much faster, in order to obtain a powerful repolarization and fAHP. Interestingly, Lien & Jonas (2003) have given a nice illustration of this point by showing directly, using dynamic clamp, that the analogous role of Kv3 current in interneurons depends on the deactivation kinetics in exactly this way. C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 878 N. Gu and others BK current inactivation and a novel form of spike frequency adaptation As previously described, several lines of evidence indicate that the BK current in CA1 pyramidal cells is transient, i.e. it decays within a few milliseconds during sustained depolarization (Storm, 1990; Halvorsrud et al. 1999; Shao et al. 1999a; Vervaeke et al. 2006a). Therefore, the current was dubbed ‘I CT ’, i.e. transient Ca2+ -activated K+ current, by Storm (1990) and Shao et al. (1999a). Our group has previously also shown how this inactivation produces frequency-dependent spike broadening and rapid decay of the fAHP during a high-frequency discharge (Shao et al. 1999a). The inactivation of the BK current will also necessarily influence excitability and spike frequency, in particular spike frequency adaptation. As outlined above, the spike frequency is reduced when I BK disappears, due to enhanced I DR activation and Na+ channel inactivation causing enhanced refractoriness. Therefore, the inactivation of I BK during the initial spikes in a high-frequency train should contribute to spike frequency adaptation. This logical, qualitative prediction was confirmed by our modelling as well as our experiments. For example, in Fig. 8A, the third ISI is virtually identical with BK current (black, continuous line: 130 pS μm−2 ) and without (red line), whereas the first ISI is about 30% longer when the BK current is missing, indicating reduced adaptation. A similar effect was seen experimentally in response to IbTX (Fig. 2B). Figure 8A also shows that the inactivation of I Na (I Na I-state) increased following the 3rd spike compared with the firsts spike with normal BK current (black line), whereas there is no such increase without BK channels (red line; actually the I-state here is slightly reduced). Thus, because the BK current facilitates spiking but inactivates rapidly, it contributes to early spike frequency adaptation. Interestingly, this mechanism of I BK -dependent spike frequency adaptation is almost exactly the opposite to the ‘classical’ mechanism of K+ channel-dependent spike frequency adaptation, which has been described for, e.g. the ‘Ks’ current in molluscan neurons (Partridge & Stevens, 1976), the M- and SK-currents in sympathetic neurons (Adams et al. 1986), the M- and sAHP-currents in hippocampal and neocortical neurons (Madison & Nicoll, 1984; Storm, 1989; Gu et al. 2005; Peters et al. 2005). In all the latter cases, the adaptation results from a cumulative build-up of a slowly activating K+ conductance, causing a progressive reduction in excitability and spike frequency, often in the form of a true feed-back regulation of spike frequency (Partridge & Stevens, 1976; Storm, 1990; Hille, 2001). In contrast, in the I BK -dependent spike frequency adaptation, the K+ conductance that is the primary cause of the adaptation, g BK , enhances high-frequency firing rather than slowing it down, and this K+ conductance is decaying rather than building up. Only secondarily, J Physiol 580.3 as a consequence of g BK inactivation and the resulting spike broadening, is there an increased recruitment of other, slower K+ conductances (mainly g DR ), but this occurs by an increase in K+ conductance activation by each (broadened) spike, rather than the cumulative build-up of slowly deactivating K+ conductances, which characterizes the classical cases of K+ current-dependent adaptation, e.g. I Ks , I M , I Ks , I SK /I AHP , and I sAHP (Partridge & Stevens, 1976; Adams et al. 1986; Madison & Nicoll, 1984; Storm, 1990; Gu et al. 2005). Thus, I BK -dependent adaptation is due to a selective K+ conductance-dependent facilitation of the early spikes rather than a selective K+ conductance-dependent reduction of the later spike frequency. To our knowledge, this type of adaptation mechanism has not previously been described. Comparison with previous results In view of the widespread idea that K+ channels are mainly inhibitory, serving to limit excitation (e.g. Connor & Stevens, 1971; Partridge & Stevens, 1976; Madison & Nicoll, 1982; Madison & Nicoll, 1984; Storm, 1990; Hille, 2001), it is perhaps not surprising that previous reports mention only the inhibitory role of BK channels (Lancaster & Nicoll, 1987; Alger & Williamson, 1988; Williamson & Alger, 1990; Storm, 1990), and the same limitation applies to both early and recent reviews of BK channels and their functions (Blatz & Magleby, 1987; Marty, 1989; Sah & McLachlan, 1992; Vergara et al. 1998; Salkoff et al. 2006). Thus, based on the report that BK channel blockade increased the initial discharge frequency and reduced burst duration in CA1 pyramidal cells, it was previously suggested that the effect of BK channels is to reduce the spike frequency and excitability (Lancaster & Nicoll, 1987; Alger & Williamson, 1988; Williamson & Alger, 1990). In apparent agreement, subsequent studies suggested that BK channels contribute significantly to the medium AHP following a spike burst, in particular when the cell was depolarized (Williamson & Alger, 1990). Other evidence also suggested that these channels may generate a small mAHP component (in current clamp) and a corresponding tail current (in voltage clamp) under artificial conditions (when several other K+ currents had been suppressed by 40 μm carbachol, etc.; Lancaster & Adams, 1986; Storm, 1989). These data were interpreted as support for an I BK component of the mAHP and early spike frequency adaptation (Lancaster & Nicoll, 1987; Storm, 1990; Williamson & Alger, 1990). As discussed above, our data clearly support the conclusion that I BK contributes to early spike frequency adaptation, although the mechanism we find is very different from the one proposed originally. However, our observation that both the selective (IbTX) and nonselective (TEA) BK channel blockers consistently inhibited the high-frequency discharge (Figs 1–3), indicates that the C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 J Physiol 580.3 BK channels enhance high-frequency firing and adaptation dominating effect of BK channels in CA1 pyramidal cells is facilitating. This result does not, however, preclude that I BK may contribute a small component to the mAHP. (However, unpublished data suggest that BK channels do not contribute appreciably to the mAHP. This is the theme of a separate study; N. Gu & J. F. Storm, in preparation). Although there are slight differences in the experimental conditions between this study and those of Lancaster & Nicoll (1987) and Williamson & Alger (1990), they all used essentially the same approach (sharp electrode intracellular recordings from CA1 pyramidal in rat hippocampal slices at 20–31◦ C, etc.). Why, then, did Lancaster & Nicoll (1987) find a reduced spike frequency after blocking the BK channels whereas we consistently found the opposite? Two factors may help to explain the apparent discrepancy. (1) We observed the strongest effects of IbTX at frequencies higher than ∼50 Hz (Fig. 5), whereas Lancaster & Nicoll (1987) limited their tests of BK channel blockade (by ChTX or TEA) to spike frequencies of ∼50 Hz or less (see their Figs 3 and 8), and ChTX was tested in only two cells. Therefore, a facilitating effect of I BK might have been overlooked. (2) Their and other early studies relied on channel blockers and manipulations (TEA, ChTX, Mn2+ , Cd2+ , Ca2+ -free medium) that are not selective for BK channels, because selective blockers were not available at the time (Lancaster & Nicoll, 1987; Storm, 1989; Williamson & Alger, 1990). Thus, the effects might have been partly due to other channels than BK. However, this explanation may not apply to ChTX (Lancaster & Nicoll, 1987; Williamson & Alger, 1990), since no other ChTX-sensitive channels than BK have been shown to exist in rat CA1 pyramidal cells. Thus, the ChTX-sensitive Kv1.3 channels do not appear to be significantly expressed in this cell type (Beckh & Pongs, 1990). The early results of Williamson & Alger (1990) and Storm (1989) may have been influenced by other factors. (i) As was common at that time, KCl-filled microelectrodes, which cause artificially high intracellular Cl− concentration, were used to achieve good voltage-clamp recordings (Storm, 1989; Williamson & Alger, 1990). Since both muscarinic agonists and high [Cl− ]i may affect multiple channel types (Akaike & Sadoshima, 1989; Zhang et al. 1994), the results obtained under those conditions are probably less representative of the physiological state than the present results, which were obtained with highly selective channel blockers and intracellular media that are likely to cause minimal perturbations (Zhang et al. 1994). (ii) Since a selective M-channel blocker was unavailable until XE991 was introduced in 1998 (Wang et al. 1998), a high dose of the cholinergic agonist carbachol (40 μm) was used to first eliminate I M (and I sAHP ), in order to study the putative BK channel contribution in relative isolation (see Fig. 4 in Storm, 1989). However, this strong cholinergic stimulation may also have affected the BK channels or the 879 Ca2+ transients that activate them. Therefore, the results from those previous studies do not really contradict the results of our present study. In conclusion, the available evidence seems to support the conclusion that BK channels facilitate high-frequency discharge in CA1 pyramidal cells under normal conditions. Comparison with other cell types Although K+ channels are often portrayed as inhibitory, it has been known for decades that fast spike repolarization caused by K+ currents is important for high-frequency firing (Hodgkin & Huxley, 1952; Hille, 2001). More recently, it has been described how channels of the Kv3 family (mainly Kv3.1 and Kv3.2), due to their unusually fast activating and deactivating properties, support sustained high-frequency spike trains, by producing brief spikes and fast deep afterhyperpolariztions (AHPs) (Rudy et al. 1999). For example, blockade of Kv3 channels in interneurons produced cumulative Na+ channel inactivation, causing slowing of the firing rate (Erisir et al. 1999; Lien & Jonas, 2003). In the light of these studies, it is not surprising that BK channels, by causing relatively fast spike repolarization and fAHP in pyramidal cells, can facilitate high-frequency firing. Since BK channels contribute to spike repolarization and fAHPs in many neurons (Adams et al. 1982; Storm, 1987a; Lancaster & Nicoll, 1987; Schwindt et al. 1988; Womack & Khodakhah, 2002), it is likely that their facilitating effect is a widespread phenomenon. This is supported by a recent paper, which appeared while this study was in progress (Brenner et al. 2005). It reports that BK channels in mice lacking BK β 4 -subunits promote repetitive firing in dentate granule cells by narrowing the spike, thus reducing the duration of the AHP. The authors also suggest that this effect, which resembles the one we report here, is a likely cause of the temporal lobe seizures of this mouse strain (Brenner et al. 2005). Clinical implications Recently, a familial form of human epilepsy and paroxysmal dyskinesia was found to be associated with a gain-of-function mutation in the BK channel α-subunit (Du et al. 2005). The mutation caused a ∼4-fold increase in the Ca2+ sensitivity of the channel, leading to a larger and faster BK current. It was speculated that this might cause increased brain excitability because of (1) faster spike repolarization and reduced Na+ inactivation, or (2) hyperpolarization-induced bursting as in absence seizures, or (3) hyperpolarization-induced activation of the h-current (Du et al. 2005). In view of the results presented here and those of Brenner et al. (2005), it seems likely that the first of these mechanisms is important for this form of epilepsy in humans. The results of Du et al. (2005) provide a fascinating example of how the BK channel-induced facilitation mechanisms described in C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 880 N. Gu and others this study might be of functional and clinical importance. In addition to these mechanisms, reduced excitability of GABAergic neurons (Zhang & McBain, 1995) or effects at GABAergic synapses (Goldberg et al. 2005) might underlie the epilepsy caused by the gain-of-function BK channel mutation (Du et al. 2005). References Adams PR, Constanti A, Brown DA & Clark RB (1982). Intracellular Ca2+ activates a fast voltage-sensitive K+ current in vertebrate sympathetic neurones. Nature 296, 746–749. Adams PR, Jones SW, Pennefather P, Brown DA, Koch C & Lancaster B (1986). Slow synaptic transmission in frog sympathetic ganglia. J Exp Biol 124, 259–285. Akaike N & Sadoshima J (1989). Caffeine affects four different ionic currents in the bull-frog sympathetic neurone. J Physiol 412, 221–244. Alger BE & Williamson A (1988). A transient calciumdependent potassium component of the epileptiform burst after-hyperpolarization in rat hippocampus. J Physiol 399, 191–205. Beckh S & Pongs O (1990). Members of the RCK potassium channel family are differentially expressed in the rat nervous system. EMBO J 9, 777–782. Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U & Fakler B (2006). BKCa-Cav channel complexes mediate rapid and localized Ca2+ -activated K+ signaling. Science 314, 615–620. Blatz AL & Magleby KL (1987). Calcium-activated potassium channels. Trends Neurosci 10, 463–467. Borg-Graham L (1987). Modelling the somatic electrical behaviour of hippocampal pyramidal neurons. Master’s Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA (also appears at MIT AI laboratory Technical report 1161) Ref Type: Thesis/Dissertation. Borg-Graham L (1999). Interpretations of data and mechanisms for hippocampal pyramidal cell models. In Cerebral Cortex, Vol. 12: Cortical Models, ed. Jones EG, Ulinski PS & Peter A. Plenum Press, New York. Borg-Graham LJ (2000). Additional efficient computation of branched nerve equations: adaptive time step and ideal voltage clamp. J Comput Neurosci 8, 209–226. Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL & Aldrich RW (2005). BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci 8, 1752–1759. Brown DA & Adams PR (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283, 673–676. Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P & Litt M (1994). Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 8, 136–140. Cai X, Liang CW, Muralidharan S, Kao JP, Tang CM & Thompson SM (2004). Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 44, 351–364. J Physiol 580.3 Connor JA & Stevens CF (1971). Prediction of repetitive firing behaviour from voltage clamp data on an isolated neurone soma. J Physiol 213, 31–53. Du J, Haak LL, Phillips-Tansey E, Russell JT & McBain CJ (2000). Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol 522, 19–31. Du W, Bautista JF, Yang H, ez-Sampedro A, You SA, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Richerson GB & Wang QK (2005). Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet 37, 733–738. Erisir A, Lau D, Rudy B & Leonard CS (1999). Function of specific K+ channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J Neurophysiol 82, 2476–2489. Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ & Garcia ML (1990). Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem 265, 11083–11090. Gho M & Ganetzky B (1992). Analysis of repolarization of presynaptic motor terminals in Drosophila larvae using potassium-channel-blocking drugs and mutations. J Exp Biol 170, 93–111. Gola M & Crest M (1993). Colocalization of active KCa channels and Ca2+ channels within Ca2+ domains in helix neurons. Neuron 10, 689–699. Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS & Rudy B (2005). Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J Neurosci 25, 5230–5235. Graham LJ (2004). The Surf-Hippo Neuron Simulation System, v3.5a Ref Type: Computer Program http://www. neurophys.biomedicale.univ-paris5.fr/∼graham/surf-hippo. html Gu N, Vervaeke K, Hu H & Storm JF (2005). Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol 566, 689–715. Gu N, Vervaeke K & Storm JF (2006). Large-conductance calcium-activated K+ channels (BK) channels promote high-frequency firing in rat CA1 hippocampal pyramidal neurons. FENS Forum Abstracts 3, A189.7. Gustafsson B, Galvan M, Grafe P & Wigstrom H (1982). A transient outward current in a mammalian central neurone blocked by 4-aminopyridine. Nature 299, 252–254. Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC & Brown DA (2000). Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br J Pharmacol 129, 413–415. Halvorsrud R, Shao LR, Bouskila Y, Ramakers GM & Storm JF (1999). Evidence that BK-type Ca2+ -dependent K+ channels contribute to frequency-dependent action potential broadening in rat CA1 hippocampal pyramidal cells. Abstr Soc Neurosci 25, 453. C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 J Physiol 580.3 BK channels enhance high-frequency firing and adaptation Harris KD, Hirase H, Leinekugel X, Henze DA & Buzsaki G (2001). Temporal interaction between single spikes and complex spike bursts in hippocampal pyramidal cells. Neuron 32, 141–149. Hille B (2001). Ion Channels of Excitable Membranes, 3rd edn. Sinauer Associates, Inc., Sunderland, MA, USA. Hines M (1984). Efficient computation of branched nerve equations. Int J Biomed Comput 15, 69–76. Hodgkin AL & Huxley AF (1952). Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol 116, 449–472. Hoffman DA, Magee JC, Colbert CM & Johnston D (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875. Hotson JR & Prince DA (1981). Penicillin- and barium-induced epileptiform bursting in hippocampal neurons: actions on Ca2+ and K+ potentials. Ann Neurol 10, 11–17. Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP & Storm JF (2001). Presynaptic Ca2+ -activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci 21, 9585–9597. Hu H, Vervaeke K & Storm JF (2002). Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol 545, 783–805. Jin W, Sugaya A, Tsuda T, Ohguchi H & Sugaya E (2000). Relationship between large conductance calcium-activated potassium channel and bursting activity. Brain Res 860, 21–28. Johnston D, Hoffman DA, Magee JC, Poolos NP, Watanabe S, Colbert CM & Migliore M (2000). Dendritic potassium channels in hippocampal pyramidal neurons. J Physiol 525, 75–81. Lancaster B & Adams PR (1986). Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J Neurophysiol 55, 1268–1282. Lancaster B & Nicoll RA (1987). Properties of two calciumactivated hyperpolarizations in rat hippocampal neurones. J Physiol 389, 187–203. Lien CC & Jonas P (2003). Kv3 potassium conductance is necessary and kinetically optimized for high-frequency action potential generation in hippocampal interneurons. J Neurosci 23, 2058–2068. Madison DV & Nicoll RA (1982). Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature 299, 636–638. Madison DV & Nicoll RA (1984). Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol 354, 319–331. Marrion NV & Tavalin SJ (1998). Selective activation of Ca2+ -activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395, 900–905. Marty A (1989). The physiological role of calcium-dependent channels. Trends Neurosci 12, 420–424. Nelson AB, Krispel CM, du Sekirnjak C & LS (2003). Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron 40, 609–620. 881 Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J & Adelman JP (2005). SK channels and NMDA receptors form a Ca2+ -mediated feedback loop in dendritic spines. Nat Neurosci 8, 642–649. Partridge LD & Stevens CF (1976). A mechanism for spike frequency adaptation. J Physiol 256, 315–332. Pedarzani P & Storm JF (1993). PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron 11, 1023–1035. Peters HC, Hu H, Pongs O, Storm JF & Isbrandt D (2005). Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 8, 51–60. Rall W, Burke RE, Holmes WR, Jack JJ, Redman SJ & Segev I (1992). Matching dendritic neuron models to experimental data. Physiol Rev 72, S159–S186. Ramakers GM & Storm JF (2002). A postsynaptic transient K+ current modulated by arachidonic acid regulates synaptic integration and threshold for LTP induction in hippocampal pyramidal cells. Proc Natl Acad Sci U S A 99, 10144–10149. Ranck JB Jr (1973). Studies on single neurons in dorsal hippocampal formation and septum in unrestrained rats. I. Behavioral correlates and firing repertoires. Exp Neurol 41, 461–531. Rudy B, Chow A, Lau D, Amarillo Y, Ozaita A, Saganich M, Moreno H, Nadal MS, Hernandez-Pineda R, Hernandez-Cruz A, Erisir A, Leonard C & Vega-Saenz de ME (1999). Contributions of Kv3 channels to neuronal excitability. Ann N Y Acad Sci 868, 304–343. Rudy B & Mcbain CJ (2001). Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci 24, 517–526. Rutecki PA, Lebeda FJ & Johnston D (1987). 4-Aminopyridine produces epileptiform activity in hippocampus and enhances synaptic excitation and inhibition. J Neurophysiol 57, 1911–1924. Sah P & Faber ES (2002). Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol 66, 345–353. Sah P & McLachlan EM (1992). Potassium currents contributing to action potential repolarization and the afterhyperpolarization in rat vagal motoneurons. J Neurophysiol 68, 1834–1841. Salkoff L, Butler A, Ferreira G, Santi C & Wei A (2006). High-conductance potassium channels of the SLO family. Nat Rev Neurosci 7, 921–931. Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H et al. (2004). Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+ -activated K+ channel deficiency. Proc Natl Acad Sci U S A 101, 9474–9478. Schwindt PC, Spain WJ, Foehring RC, Stafstrom CE, Chubb MC & Crill WE (1988). Multiple potassium conductances and their functions in neurons from cat sensorimotor cortex in vitro. J Neurophysiol 59, 424–449. Shao LR, Halvorsrud R, Borg-Graham L & Storm JF (1999a). The role of BK-type Ca2+ -dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol 521, 135–146. C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010 882 N. Gu and others Shao LR, Halvorsrud R, Bouskila Y, Ramakers GM, Borg-Graham L & Storm JF (1999b). Evidence that BK-type Ca2+ -dependent K+ channels contribute to frequencydependent action potential broadening in rat CA1 hippocampal pyramidal cells. Physiologist 42, A-21. Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A & Tempel BL (1998). Deletion of the KV 1.1 potassium channel causes epilepsy in mice. Neuron 20, 809–819. Steinlein OK (2001). Genes and mutations in idiopathic epilepsy. Am J Med Genet 106, 139–145. Stocker M, Krause M & Pedarzani P (1999). An apaminsensitive Ca2+ -activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A 96, 4662–4667. Storm JF (1987a). Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol 385, 733–759. Storm JF (1987b). Intracellular injection of a Ca2+ chelator inhibits spike repolarization in hippocampal neurons. Brain Res 435, 387–392. Storm JF (1988). Temporal integration by a slowly inactivating K+ current in hippocampal neurons. Nature 336, 379–381. Storm JF (1989). An after-hyperpolarization of medium duration in rat hippocampal pyramidal cells. J Physiol 409, 171–190. Storm JF (1990). Potassium currents in hippocampal pyramidal cells. Prog Brain Res 83, 161–187. Storm JF, Sartorius T, Gu N, Sausbier M, Sausbier U & Ruth P (2006). Alterations in cortical EEG activity, neuronal excitability and sleep-wake behavior in BK channel-deficient mice. Abstr Soc Neurosci 627.9, D23. Stuart G & Spruston N (1998). Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J Neurosci 18, 3501–3510. Swensen AM & Bean BP (2003). Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci 23, 9650–9663. Takahashi T (1990). Membrane currents in visually identified motoneurones of neonatal rat spinal cord. J Physiol 423, 27–46. Vergara C, Latorre R, Marrion NV & Adelman JP (1998). Calcium-activated potassium channels. Curr Opin Neurobiol 8, 321–329. Vervaeke K, Gu N & Storm JF (2006a). Large conductance calcium activated K+ (BK) channels promote highfrequency firing in rat CA1 hippocampal pyramidal neurons. Abstr Soc Neurosci 627.7, D21. Vervaeke K, Hu H, Graham LJ & Storm JF (2006b). Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron 49, 257–270. Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE & Mckinnon D (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. J Physiol 580.3 Williamson A & Alger BE (1990). Characterization of an early afterhyperpolarization after a brief train of action potentials in rat hippocampal neurons in vitro. J Neurophysiol 63, 72–81. Womack MD & Khodakhah K (2002). Characterization of large conductance Ca2+ -activated K+ channels in cerebellar Purkinje neurons. Eur J Neurosci 16, 1214–1222. Zbicz KL & Weight FF (1985). Transient voltage and calciumdependent outward currents in hippocampal CA3 pyramidal neurons. J Neurophysiol 53, 1038–1058. Zhang XF, Gopalakrishnan M & Shieh CC (2003). Modulation of action potential firing by iberiotoxin and NS1619 in rat dorsal root ganglion neurons. Neuroscience 122, 1003–1011. Zhang L & McBain CJ (1995). Potassium conductances underlying repolarization and after-hyperpolarization in rat CA1 hippocampal interneurones. J Physiol 488, 661–672. Zhang L, Weiner JL, Valiante TA, Velumian AA, Watson PL, Jahromi SS, Schertzer S, Pennefather P & Carlen PL (1994). Whole-cell recording of the Ca2+ -dependent slow afterhyperpolarization in hippocampal neurones: effects of internally applied anions. Pflugers Arch 426, 247–253. Acknowledgements We thank Lyle Graham, university Rene Descartes, Paris, for the continuous development and support of the Surf-Hippo simulator. This study was supported by the Norwegian Research Council (N.F.R.) via the MH, FUGE, EMBIO and Norwegian Centre of Excellence programmes. G.N. performed all the intracellular current-clamp experiments included in this study (including those presented in Figs 1–4, 5A and B, 7 and 9A–C); K.V. performed the computer modelling (illustrated in Figs 5C and D, 6, 8, 9D–F and 10) and extracellular field potential recording illustrated in Fig. 11. J.F.S. performed pilot intracellular current-clamp experiments. The manuscript was mainly written by G.N. and J.F.S. Supplemental material The online version of this paper can be accessed at: DOI: 10.1113/jphysiol.2006.126367 http://jp.physoc.org/cgi/content/full/jphysiol.2006. 126367/DC1 and contains supplemental material entitled: Supplemental computational methods (reprinted from Neuron 49, 257–270. Vervaeke K, Hu H, Graham LJ & Storm JF (2006). Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing, with permission from Elsevier). This material can also be found as part of the full-text HTML version available from http://www.blackwellsynergy.com C 2007 The Authors. Journal compilation C 2007 The Physiological Society Downloaded from J Physiol (jp.physoc.org) at UNIV OF TEXAS-AUSTIN on February 24, 2010