Synthesis 3: A Synthetic Competition
advertisement

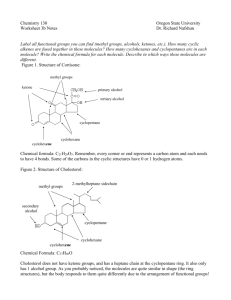
Synthesis 6: A Synthetic Competition [Taken from: Stradling, S.S.; Gage, C.L. J. Chem. Ed. 1985, 62, 1117-7] O HNO3 /H 2 SO 4 NaOCl O O acetophenone OH benzoic acid NO2 m-nitroacetophenone CH 3OH/H + O HNO3 /H 2 SO 4 NaOCl OCH 3 O methyl benzoate O OH OH HNO3 /H 2 SO 4 NO2 m-nitrobenzoic acid NO2 m-nitrobenzoic acid O CH 3OH/H + OCH 3 CH 3OH/H + NO2 methyl m-nitrobenzoate Path 1: Acetophenone Benzoic acid m-Nitrobenzoic acid Path 2: Acetophenone Benzoic acid Methyl benzoate Path 3: Acetophenone m-Nitroacetophenone Methyl m-nitrobenzoate Methyl m-nitrobenzoate m-Nitrobenzoic acid Methyl m-nitrobenzoate Introduction There is often more than one way to synthesize a particular product from a given starting material. In the scheme above, three methods are shown for synthesizing methyl mnitrobenzoate starting with acetophenone. Your goal in this experiment is to have a competition with two other people to determine which is the most cost-efficient path for the synthesis. Your economic cost depends on the amounts of each chemical you use and the purity of your final product! The chemical costs can be found in the Aldrich catalog. You will not be charged for water or utilities, but you can calculate a cost for person hours. Your overall grade will be determined by three factors: quality of analysis of product, yield of product, and purity of product. A great write up of a low purity low yield will not be good! Experimental FOR THIS LAB YOU WILL HAVE THREE LAB PERIODS TO MAKE THIS TARGET MATERIAL WITH ONE DAY OF WRAP-UP BEFORE SPRING BREAK. Everyone starts with 8.0 mL acetophenone. The procedures listed below are the same regardless of the particular step you are on. You need to decide if you want to purify things after each step since that will cost you. Conversion of a Methyl Ketone to a Carboxylic Acid. Place your weighed methyl ketone in an 800-mL beaker with 40 mL of 5% NaOCl (common bleach) and 2.5 mL of 10% NaOH per one gram of methyl ketone (both reagents per 1 g methyl ketone!). Warm and stir (not boil!) the contents of the beaker for 20-30 min. Slowly add 4 mL acetone to destroy excess NaOCl. Cool the mixture slightly and add concentrated HCl slowly with stirring until the pH is 2-3. Cool in an ice bath and vacuum filter the product. Characterize the product and/or purify it. Preparation of a Methyl Ester from a Carboxylic Acid. Set up a reflux apparatus with an appropriate round bottom flask. Add your carboxylic acid and 8 mL of methanol per one gram of carboxylic acid. Add 1 mL of concentrated sulfuric acid per 20 mL methanol some boiling stones and reflux for ~1 h. It is important that your starting material be dry. The workup steps that follow are different since one product is a solid and the other a liquid. If you are making methyl m-nitrobenzoate, pour the reaction mixture over ice water (use about 5 times the volume of alcohol used), stir, and allow the ice to melt. Vacuum filter the product and characterize and/or purify it. If you are making methyl benzoate, pour the reaction mixture onto a handful of ice and add 10% NaOH until the solution is basic. Extract the aqueous solution twice with ether (use a volume of ether for each extraction equal to one-half the volume of alcohol used). Dry the ether layer, filter off the drying agent, and evaporate off the ether on the rotovap. Isolate the product by short-path distillation. Synthesis of m-Nitroacetophenone Prepare a nitrating mixture by slowly adding 8 mL of concentrated H2SO4 to 5.3 mL of concentrated HNO3 in a small Erlenmeyer flask in an ice bath. Cool this mixture to 0°C. Place 20 mL concentrated H2SO4 in a 500 mL Erlenmeyer flask and cool to 0°C with swirling. Add the 8.0 mL acetophenone dropwise to the cold H2SO4 at such a rate that the temperature does not exceed 5°C. Cool this mixture to below 0°C in an ice/salt bath (ice + NaCl will bring down the temp to ~ -50C, you will need to add at least 5 g of NaCl to your beaker containing ice and mix with stirring rod, You will need to replace this mixture if you see ice melting as time proceeds!) and add the cold nitrating mixture dropwise, keeping the temp at or below 0°C (it may take a good 15 mins for this addition step). After the addition, maintain the temperature at 0°C for another 10-15 min with occasional swirling. Pour the contents of the flask into a beaker containing ~100 g ice and ~150 mL water with vigorous stirring. When the excess ice has melted, vacuum filter the solid and wash it well with cold water (the product might be sticky!). Disconnect the vacuum and add 5 mL ice-cold ethanol to the contents of the funnel. Let it stand for about a minute and then reattach to vacuum line to finish the filtration. Spread the crude product out on some filter paper to allow it to dry. Characterize and/or purify the product. If you do not see solid product then you may need to extract the product (found in the ice/water layer) with ethyl acetate. Dry the organic layer with Na2SO4 and then rotovap off the ethyl acetate. If you do not see any solid at this point, then you did not successfully perform the nitration! Synthesis of m-Nitrobenzoic Acid and Methyl m-Nitrobenzoate For every gram of organic starting material, use 1.0 mL of H2SO4 and 0.67 ml of HNO3. Prepare the nitrating mixture by slowly adding 3 parts of H2SO4 to two parts of HNO3 with cooling. In a 500 mL Erlenmeyer flask place concentrated H2SO4 (use 2.5 mL of H2SO4 per gram of starting material). Cool to 0°C, and then add the organic substrate with stirring. The temperature should not exceed 5°C. Follow the directions in the acetophenone procedure above, but do NOT wash with cold ethanol. Characterize and/or purify the product.