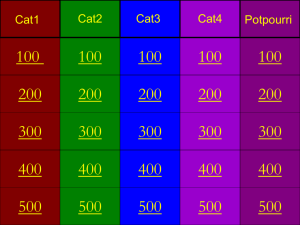
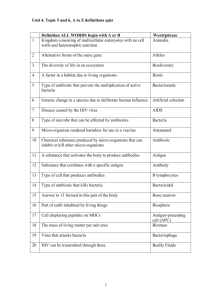
Table of Contents
advertisement

Table of Contents Introduction to the concept of selected writing in Microbiology…………………………………………….. Page 1 Writing a paper about a disease or an infectious agent………………………………………………………….. Page 2 Rubric for Grading Research papers……………………………………………………………………………………….. Page 3 Writing Lab Reports and Scientific Papers………………………………………………………………………………. Page 4 Rubric for Grading Laboratory reports……………………………………………………………………….. Page 8 Section 1: Group Projects Comparison of the effectiveness of Ticlosan antibacterial soap and regular soap on Gram-positive and Gram-negative bacteria ………………………………………………………………………….. Page 10 Comparison of Endospore Staining Techniques Using Bacillus cereus……………………………………. Page 15 Bacteriological Examination of Salad: Comparing Bacterial counts ………………………………………. Page 18 Comparison of Science Building with older buildings for levels of contamination of coliforms in male and female lavatories…………………………………………………………………………………………………. Page 22 Determination of bacterial contamination of public computer keyboards……………………………. Page 26 Microbiological evaluation of quality of drinking water at Lincoln university………………………. Page 29 The Effectiveness of Alcohol based and Non- Alcohol based Mouthwashes against Staphylococcus aureus and Pseudomonas aeruginosa………………………………………………………… Page 33 Section 2: Research Papers Syphilis…………………………………………………………………………………………………………………………………. Page 38 Bovine spongiform encephalopathy …………………………………………………………………………………… Page 44 HERPES: A Sexually Transmitted Disease…………………………………………………………………………….. Page 49 Borrelia burgdorferi……………………………………………………………………………………………………………… Page 53 i Malaria ……………………………………………………………………………………………………………………………….. Page 59 Influenza……………………………………………………………………………………………………………………………… Page 64 Clostridium botulinum…………………………………………………………………………………………………………. Page 70 Human Immunodeficiency Virus (HIV)……………………………………………………………………………………… Page 75 Anthrax: an acute disease caused by Bacillus anthracis………………………………………………………….. Page 82 Section 3: Laboratory Reports Determination of the growth curve of Escherichia coli in batch culture………………………………….. Page 88 Isolation of Klebsiella pneumoniae and Enterobacter aerogenes colonies and examination of morphological characteristics…………………………………………………………………………………………………… Page 92 Simple and Gram staining of Klebsiella pneumoniae and Enterobacter aerogenes………………….. Page 94 Evaluation of Pathogen Sensitivity to Chemotherapeutic Agents…………………………………………….. Page 97 Determination of Water Quality by Presumptive, Confirmed, and Completed Tests…………… Page 100 Staining of Mycobacterium smegmatis for acid fastness and Bacillus cereus for endospores….. Page 103 ii Introduction to the concept of selected writing in Microbiology When I began teaching Microbiology at Lincoln University in 1995, I quickly realized that many students had difficulty with writing in the sciences. Specifically, students had problems with referencing styles, reporting laboratory experiments and writing research papers. Over the years, I have therefore, used my teaching time to encourage students to write. In order to encourage students to be enthusiastic about writing, I have adopted a non-punitive attitude in which drafts are given feedback and only the final submission is graded. This approach has enabled my students to view the exercise as an opportunity to improve their writing skills, and has also encouraged them to take the feedback more seriously than they would have if I had graded the first draft. I also provide writing formats for laboratory reports and research papers to students along with rubrics for their grading. This approach has not only cut down on the copy and paste syndrome, but also restricted students to a disciplined approach to writing. Over the years, I have used three learning opportunities for writing assignments. Firstly, there is usually an average of eight laboratory exercises in a semester; therefore, all laboratory reports are written to mimic submissions of scientific laboratory studies. The rationale is that if students can write four or five good scientific reports, they would be more likely to adopt the habit of writing in that format in the future. That future could be summer internships or graduate or professional school experience. Secondly, students ought to know how to write a research paper. I let students accomplish this goal by writing about a disease or an infectious agent. A format is also provided that mimics that of a review article normally seen in Journals of Microbiology or Infectious disease. Students are made aware of how to make writing about an obscure disease or infectious agent relevant as we discuss their significance, symptoms and pathogenicity. Lastly, students are made to write about joint or group projects. In this effort, I inculcate in students the ability to recognize problems, ask research questions, design study protocols to answer the research questions and report their findings in a scientific format. These approaches are by no means the only ones for encouraging students to write in the discipline. However, they have always worked for me because they keep the students focused throughout the semester. They also make students quickly recognize the practical or application side of what they have been learning in theory. Since 1997, I have made it a practice to collect students’ writings and publish them whenever I can. This is helpful to students as they realize that their writings are important even to the professor and other audiences. I have been informed by some of my students that their job offers were made possible because of showing the published writings to their potential employers. In the Spring of 2010, I taught the General Microbiology course and have selected some of the students’ writings for publication. There are group projects, research papers and laboratory reports. The reports have not been significantly altered during the editing process, and therefore, represent the writing efforts of the students. We have a long way to go to get our students to write professionally; this is just a beginning. I hope that you will enjoy reading my students’ works. John O. Chikwem 1 Writing a paper about a disease or an infectious agent Introduction: Writing about diseases or infectious agents is part of the work carried out by microbiologists. All research involving infectious agents or the diseases that they cause must be covered according to the following headings: General description of the agent or the disease. if you are writing about the bacterium, Staphylococcus aureus, you will describe it as a Gram positive, coagulase positive cocci that appear in clusters and are golden yellow on Nutrient agar, etc. On the other hand, if you are writing about the disease gonorrhoea, you will describe it as a sexually transmitted infection caused by the bacterium Neisseria gonorrhoeae. It produces inflammation and infection of the lining of the genital tract, throat, and rectum, etc. The goal is to describe the agent or the disease is a way that those familiar with it can easily recognize it. Significance: In this section, you need to discuss why this organism or disease is important and what makes it important. A disease or infectious agent may be important because it kills a lot of people, or affects a lot of people, or produces a lot of complications or costs a lot of money to treat or is difficult to diagnose, etc. When you discuss this area, you need to use data effectively in order to show that this disease or infectious agent is worthy of the discussion. General Symptoms: In this section you will need to inform your reader how they can recognize this illness or infection caused by the agent. For example, how long does it take from the first contact with the agent to the time the symptoms manifest and what are those symptoms? Are symptoms different in males and females, adults and infants? Pathogenesis: In this section, you will need to inform your reader about what is known about the mechanism of disease causation. Every infectious agent causes disease because it has something that enables it to overpower or evade the immune system. For example, the three major causative agents of bacterial meningitis – Streptococcus pneumoniae, Neisseria meningitidis and Haemophilus influenzae are all capsulated. The capsules enable them to evade phagocytosis. Laboratory Diagnosis: In this section you are required to discuss the types of specimens required for diagnosis, how they should be collected, when they should be collected and what type of tests are required. You will also discuss the results that will confirm diagnosis. Treatment: This usually involves the use of chemical agents to get rid of the infectious agent or disease symptoms. Duration of treatment should also be discussed. Prevention: This entails discussing the usual personal strategies to avoid infection or the disease. This section may involve hygiene or avoiding infected persons, etc. It also involves the use of prophylactic agents like vaccines or chemicals. References: See below for lab reports and scientific papers. 2 Rubric for Grading Research papers Max Points 5 10 20 10 15 10 10 10 10 100 Sections 0.0 0.25 Title: Paper has title, author’s name and course. Title describes content concisely, adequately & appropriately. Description of infectious agent or disease: Characteristics such as size, shape, Gram reaction, growth characteristics of microbes, etc . For diseases, features of disease are adequately described. Significance: Data on numbers of people affected each year including deaths; Examples of outbreaks or epidemics; complications of infections; losses due to infection or disease including hospitalization costs, lawsuits, recalls and other losses. Symptoms: Incubation period, typical symptoms after infection; symptoms during complications. Pathogenesis: What mechanisms enable the organism to cause disease? Example: invasiveness, toxin production, etc Laboratory diagnosis: Samples required; Media for cultivation of samples; Other tests for identification including serology and biochemical tests. Treatment: Antibiotics used for treatment; other strategies including rehydration, treatment of complications, etc. Prevention: Hygiene and aseptic techniques; quarantine, contact tracing, vaccination, prophylaxis, etc. References: References follow Vancouver style; current; cited in text; Journal articles included. Total points 0.0 (Very poor); 0.25 (poor); 0.5 (Fair); 0.75 (Good); 1.0 (Excellent) 3 0.5 0.75 1.0 Points Writing Lab Reports and Scientific Papers Many scientists work in various laboratories and perform all sorts of experimental procedures; however, not very many are able to communicate their findings to others in writing. Therefore, the results and conclusions that were derived from such experiments are of little scientific value since they were not effectively communicated to others. In the academic field, many instructors have earned Ph.Ds; however, not many attain tenure in research intensive institutions due to the fact that they do not have sufficient collection of published scientific work. Even those who are not university scientists need to write reports of their work in order to preserve the scientific integrity of their discoveries and to show their contributions in the workplace. In a busy workplace, the boss may not know the worker, but nevertheless, must make decisions involving promotions, layoffs, terminations, etc. Those workers who have sufficient records of written reports have a greater advantage over others irrespective of the volume of work performed. Writing a good laboratory report is therefore an excellent way to start your development into an effective scientific technical writer. A good laboratory report should have a structured format. The following format is utilized by all scientists who submit materials to journals for publication. I therefore expect that all laboratory exercises should be reported as follows: 1. 2. 3. 4. 5. 6. 7. Title Abstract Introduction Materials and methods Results Discussion Literature cited Title The title should be about ten words and should convey the factual content of the paper. A good title is straightforward and uses keywords that researchers in a particular field will recognize. A good rule is to write all the key words involved in the exercise, and then string them together in a way that makes sense. Remember not to make your title too general because if the report is published, it may be difficult to locate during a web-search. After the title, please include your name and that of your laboratory partner. It is usual to add addresses of the authors; however, since this is a class exercise, no address of authors will be added. In the place of address, we will just include the Course title, semester and year. Example: Isolation of bacterial colonies and examination of morphological characteristics John O. Chikwem General Microbiology, Spring 2010 4 Abstract There are many scientific manuscripts published each year; however, we are so busy that we cannot read all of them. The abstract allows the reader to quickly decide whether it would serve his or her interest to read the entire report. For example, there are thousands of shops that sell shoes in Philadelphia or New York, and if a shopper had only very little time to shop for shoes, it would be impractical to physically enter every shop to look for the item. A good abstract is like the window dressing that compels the shopper to enter the shop, with the likelihood that the shoe of choice would be found. A good abstract is concise (100 to 200 words), and is a summary of what was done, how it was done, the most important results and the major conclusions from the results. Introduction The introduction defines the subject of the report. It must outline the scientific purpose(s) or objective(s) for the research performed and give the reader sufficient background to understand the rest of the report. Care should be taken to limit the background to whatever is pertinent to the experiment. Scientists do not perform studies just for the sake of it. We usually have an observation that compels us to pose a research question. Therefore the introduction must answer several questions, including the following: Why was the study performed? Answers to this question may be derived from observations or from the literature. What information already exists about the study? In order to answer this question, you must review the literature, showing the historical development of the study and including the confirmations, conflicts, and gaps in existing knowledge that justify further study. What is the specific purpose of the study? The specific hypothesis and experimental design pertinent to investigating the topic should be described. Materials and Methods A good methods and materials section is like writing a recipe for a cookbook. It must therefore be written with the intention of providing enough detail for the reader to understand the experiment without overwhelming him or her. When procedures from a lab book or another report are followed exactly, simply cite the work, noting that details can be found in that particular source. However, it is still necessary to describe special pieces of equipment and the general theory of the assays used. The reader may want to duplicate your experiment, so, every detail must be provided to enable this duplication to take place. However, the writing must still be professionally done to avoid providing unnecessary routine details. Generally, this section attempts to answer the following questions: 5 What materials were used? How were they used? Where and when was the work done? (This question is most important in field studies.) Results The results section should summarize the data from the experiments without discussing their implications. The data should be organized into tables, figures, graphs, photographs, and so on. All figures and tables should have descriptive titles and should include a legend explaining any symbols, abbreviations, or special methods used. Figures and tables should be numbered separately and should be referred to in the text by number. Figures and tables should be self-explanatory, and all columns and rows in tables and axes in figures should be labeled. This section of the report should concentrate on general trends and differences and not on trivial details. Discussion This section should not just be a restatement of the results but should emphasize interpretation of the data, relating them to existing theory and knowledge. Suggestions for the improvement of techniques or experimental design may also be included here. In writing this section, you should explain the logic that allows you to accept or reject your original hypothesis. You should also be able to suggest future experiments that might clarify areas of doubt in your results. Literature Cited This section lists all articles or books cited in your report. Different journals require different formats for citing literature. For scientific writings we use the Harvard and Vancouver styles of referencing. For all laboratory reports in this course, we will use the Vancouver style of referencing. Here, the articles are number-cited as they appear. Vancouver system examples: 1. Chikwem [1] has argued that…………. 2. Studies have shown that HIV is transmissible through unprotected sex, [1] by sharing needles and syringes [3] and by an infected mother to her child during pregnancy, [4] childbirth [5] and breastfeeding.[6] 1. Citing a Journal article: Name(s) of author(s). Title of article. Title of Journal. Year of publication; Volume number: Page numbers. Chikwem JO and Royer, D. Effect of euthanasia on mood of field frogs. J. Mol. Biol. 2008; 12 (5): 127 – 129. 6 2. Citing a book: Name(s) of authors. Title of publication; Edition. Place of publication: Publisher; Year of publication Page numbers if applicable. Chikwem JO, Royer D, editors. The flying birds of Nigeria; 3rd Edition. Lagos (Nigeria): Citadel Press; 2006 (page number if necessary). 3. Citing a chapter or article in a book: Author(s) of chapter. Title of chapter. In: Editor(s) name, editors. Title of book. Place of publication: Publisher; Year of publication. Page numbers. Chikwem JO and Bhat K. Viral mechanisms of pathogenicity. In: Royer D and Safford S, editors. Pathogenic viruses. Oxford: Citadel Press; 2006. p. 124 – 142. 4. Citation from World Wide Web: Author(s). Title (online). Year (cited year, month and day). Available: URL: Swinton, DJ. Spectroscopic study of proteins (online). 2006 (cited 2006 May 9). Available from: URL: http://www.nih.gov/medline/news_1280.html 7 Rubric for Grading Laboratory reports Max Points 5 0.0 Sections Title Describes lab content concisely, adequately & appropriately 20 Abstract Conveys a sense of the full report concisely and effectively Introduction Successfully establishes the scientific concept of the lab. Effectively presents the objectives and purposes of the lab States hypothesis and provides logical reasoning for it. 15 Methods & Materials Gives enough details to allow for replication of procedure 15 Results Opens with effective statement of overall findings Presents visuals clearly and with sufficient support Successfully integrates verbal and visual representations Discussion Opens with effective statement of support for the hypothesis Backs up statement with references to appropriate findings Provides sufficient & logical explanation for the statement Sufficiently addresses other issues relevant to the lab 15 15 5 10 Presentation Citations and references adhere to proper format Format for tables and figures is correct Report is written in scientific style: clear to the point Grammar and spelling are correct Overall aims: The Student………. Has successfully learned what the lab is designed to teach Demonstrates clear and thoughtful scientific inquiry Accurately measures and analyzes data for lab findings 100 0.0 (Very poor); 0.25 (poor); 0.5 (Fair); 0.75 (Good); 1.0 (Excellent) 8 0.25 0.5 0.75 1.0 Points Section 1: Group Projects 9 Comparison of the effectiveness of Ticlosan antibacterial soap and regular soap on Gram-positive and Gram-negative bacteria Julia Greenfield, Stephanie Rand, Danielle McKnight, Ndubisi Chikwem, Tracey Coleman and Joey Toto General Microbiology, Spring 2010 ABSTRACT The antimicrobial efficacy of three triclosan containing antimicrobial soaps (Antibacterial Careone Hand Soap, Dial Antimicrobial Hand Soap and Soft Soap Antibacterial Hand Soap) was compared with the same brands of triclosan-free soaps (Careone Hand Soap, Dial Hand Soap and Soft Soap Hand Soap). Dilutions of the soaps were used in the preparation of pour plates which were then streaked with Escherichia coli, Pseudomonas aeruginosa, Salmonella typhimurium, Bacillus cereus, and Staphylococcus aureus. After incubation at 37oC for 18 hours, none of the regular hand soaps inhibited the growth of the bacteria. On the other hand, all the triclosan-containing antibacterial soaps inhibited the growth of the test organisms except Psudomonas aeruginosa. We can therefore conclude that triclosan-containing antibacterial hand soaps are more effective in inhibiting the growth of Gram-positive and Gram-negative bacteria. INTRODUCTION Triclosan (2,4,4’trichloro-2’-hydroxydiphenyl-ether), a broad-spectrum antimicrobial agent, has become a popular ingredient in domestic antimicrobial products as an antisepsis and anti-plaque agent. [1] Triclosan is present in antimicrobial soaps, toothpastes, mouthwashes, cosmetics, deodorants, first aid products and many more commonly used household items. While triclosan is widely available for domestic use, the chlorophenol product has been identified as an endocrine disrupter, leading to the possibility of interference with the function of thyroid hormone and estrogen. [2] The phenol component of triclosan has the potential to topically cause skin irritation, deactivate sensory nerve endings, and internally cause cold sweats, circulatory collapse, convulsions, coma and death. [3] Laboratory studies have indicated that triclosan is toxic to normal liver enzymes, and the antimicrobial has been linked to hypersensitivity reactions including eczema, allergies and asthma. [4] The indiscriminate use of triclosan in everyday use also has implications in the development of bacterial resistance; consequently, some microbiologists predict an imminent increase in antimicrobial resistance to triclosan. [5] As a result of the above concerns, the Environmental Protection Agency (EPA) has categorized triclosan as a pesticide, rating it as both an environmental and human health risk. [3] While an FDA advisory board deemed triclosan an “unacceptable health and environmental risk”, the FDA has yet to ban the additive in the United States. [4] Despite increased awareness of the potential risk associated with triclosan, it continues to be used as an additive, in common household products including soap, toothpastes, cosmetics, deodorants, aftershaves, first aid kits, clothes and toys. Although opponents of triclosan use claim that “there is little independent scientific evidence of efficacy”, *1+ the widespread use of triclosan as an antimicrobial agent is not without foundation. In this lab, the antimicrobial efficacy of three triclosan containing antimicrobial soaps (Antibacterial Careone Hand Soap, Dial Antimicrobial Hand Soap and Soft Soap Antibacterial Hand Soap) was compared with the same brands of triclosan free soaps (Careone Hand Soap, Dial Hand Soap and Soft Soap Hand Soap). Our hypothesis is that the non-triclosan containing hand soaps are equally effective in killing bacteria as the triclosan-containing hand soaps. 10 METHODS AND MATERIALS 32 grams of Veal Infusion Agar was mixed with 800 ml of distilled water in a 1000 ml Erlenmeyer flask. The mix was heated over a Bunsen burner and carefully swirled until the Veal Infusion Agar had completely dissolved into the solution. A 20 ml pipette was used to transfer 19 ml of media into each of 30 screw-cap tubes. The tubes were autoclaved at 121oC for 15 minutes. After the tubes were removed from the autoclave, they were placed in a 45oC warm water bath to prevent solidification of the media. While the tubes were in the water bath, dilutions of soap solutions were made to give stock solutions of 10.0, 5.0, 2.0, 1.0, and 0.5 percent. 1.0 ml of each soap dilution was added to the 19 ml of molten and cooled agar. The soap dilutions and molten agar were gently mixed together to give a final soap concentration of 0.5, 0.25, 0.1, 0.05 and 0.025 percent. The agar and soap solutions were then gently poured into Petri dishes and allowed to solidify. In addition, a control agar was prepared which contained no soap. Cultures of Escherichia coli, Pseudomonas aeruginosa, Salmonella typhimurium, Bacillus cereus, and Staphylococcus aureus were inoculated into tubes of Veal Infusion Broth and incubated at 37°C overnight. The broth culture of each bacterium was diluted in saline to McFarland 0.5. The soap infused agar and control agar were then inoculated with a loop-full of each culture. The five bacteria samples were streaked on each agar plate in parallel rows. Plates were incubated overnight at 37:C. Microbial growth was observed the following day and results were recorded. RESULTS Table 1: Prices and amount of triclosan in soap Brand Careone Hand Soap Careone Hand Soap Antibacterial Dial Hand Soap Dial Gold Hand Soap Antibacterial Soft Soap Hand Soap No Soft Soap Hand Soap Antibacterial Antibacterial No Yes; 0.115% triclosan No Yes; 0.15% triclosan Volume Price ($) 221 ml 1.48 221 ml 1.48 221 ml 1.59 277 ml 1.59 221 ml 1.85 221 ml 1.85 Table 2: Antibacterial effect of Careone Hand Soap (triclosan free) Careone Hand Soap Soap concentration Organism Gram 0.5% 0.25% 0.1% 0.05% Escherichia coli G G G G Pseudomonas aeruginosa G G G G Salmonella typhimurium G G G G Bacillus cereus + G G G G Staphylococcus aureus + G G G G 0.025% G G G G G Yes; 0.15% triclosan Key: Tables 2 to 7: + = Gram positive bacteria; - = Gram negative bacteria; N= No growth; G= Growth 11 Table 3: Antibacterial effect of Dial Hand Soap (triclosan free) Organism Escherichia coli Pseudomonas aeruginosa Salmonella typhimurium Bacillus cereus Staphylococcus aureus Gram + + 0.5% G G G G G Dial Hand Soap Soap concentration 0.25% 0.1% 0.05% G G G G G G G G G G G G G G G 0.025% G G G G G Table 4: Antibacterial effect of Soft Soap Hand Soap (triclosan free) Organism Escherichia coli Pseudomonas aeruginosa Salmonella typhimurium Bacillus cereus Staphylococcus aureus Gram + + 0.5% G G G G G Soft Soap Hand Soap Soap concentration 0.25% 0.1% 0.05% G G G G G G G G G G G G G G G 0.025% G G G G G Table 5: Antibacterial effect of Careone Hand Soap Antibacterial 0.115% triclosan Careone Hand Soap Antibacterial Soap concentration Organism Gram 0.5% 0.25% 0.1% 0.05% 0.025% Escherichia coli - N N N N N Pseudomonas aeruginosa - G G G G G Salmonella typhimurium - N N N N G Bacillus cereus + N N G G G Staphylococcus aureus + N N N N N Table 6: Antibacterial effect of Dial Hand Soap Antibacterial 0.15% triclosan Dial Hand Soap Antibacterial Soap concentration Organism Gram 0.5% 0.25% 0.1% 0.05% 0.025% Escherichia coli N N N N N Pseudomonas aeruginosa G G G G G Salmonella typhimurium N N N N N Bacillus cereus + N N G G G Staphylococcus aureus + N N N N N 12 Table 7: Antibacterial effect of Soft Soap Hand Soap Antibacterial 0.15% triclosan Soft Soap Hand Soap Antibacterial Soap concentration Organism Gram 0.5% 0.25% 0.1% 0.05% 0.025% Escherichia coli N N N N N Pseudomonas aeruginosa G G G G G Salmonella typhimurium N N N N N Bacillus cereus + N N G G G Staphylococcus aureus + N N N N N The results show that all the non-triclosan containing hand soaps did not inhibit the growth of the five test organisms at the concentrations used in the study. The triclosan containing hand soaps, on the other hand, inhibited the growth of all the test bacteria except Pseudomonas aeroginosa. Escherichia coli and Salmonella typhimurium, both Gram-negative bacilli and Staphylococcus aureus, a Grampositive coccus were inhibited at all concentrations of the soaps; however, Bacillus cereus, a Gram positive bacillus was only inhibited at soap concentrations of 0.5% and 0.25%. This result was consistent for all the brands of triclosan containing hand soap. There was no significant difference in the prizes of antibacterial and regular hand soaps (Table 1). DISCUSSION All of the non-antibacterial hand soaps did not inhibit the growth of the five test organisms. This result can be attributed to the concentration of soap infused into the agar. In future investigation, the concentration of soaps used in the study will be increased in order to determine the Minimum Inhibitory Concentration (MIC) of the soaps for each test bacteria. The antibacterial hand soaps were effective in inhibiting the growth of all the bacteria except Pseudomonas aeruginosa. It would also be necessary in future to increase the concentration of the triclosan containing soaps in order to determine the minimum concentration required to kill Pseudomonas aeruginosa. The growth of B. cereus was inhibited at soap concentrations of 0.25% and above, yet below this concentration of soap, bacterial growth was observed. This could mean that this bacterium is more resistant to the soaps than S. typhimurium, Staphylococcus aureus and Escherichia coli. All the sensitive test organisms had identical results in spite of the concentration of triclosan in the antibacterial soap. Since the Careone antibacterial soap had the lowest triclosan concentration of 0.115%, this result may indicate a threshold of effectiveness for triclosan; therefore, antibacterial soaps need not have more than 0.115% triclosan in order to be effective. Given that there is a growing concern regarding the widespread use of triclosan, a lower concentration in hand soaps and other products could help to allay consumer fears. Overall, hand soaps that did not include triclosan did not inhibit the growth of the bacteria, while soaps containing triclosan inhibited the growth of all test bacteria except Pseudomonas aeruginosa. This result does not support our hypothesis that non-triclosan containing hand soaps are equally effective in 13 killing bacteria. In spite of this, our results also show that the concentration of triclosan could be reduced without affecting the antibacterial property of hand soaps. REFERENCES 1. Braid JJ and Wale MCJ. The antibacterial activity of triclosan-impregnated storage boxes against Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, Bacillus cereus and Shewanella putrefaciens in conditions simulating domestic use. Journal of Antimicrobial Chemotherapy. 2002; 49: 87-94. 2. Inglis A. Antibacterial Products Aren’t Just Useless—They Can Be Killers. [Online] [May 5,2009] [Accessed April 16, 2010]. Available from: http://www.healthiertalk.com/antibacterial-productsaren-t-just-useless-they-can-be-killers-0595 3. McGhee MA. Triclosan. [Online] [Accessed April 16, 2010]. Available from: http://www.healthreport.co.uk/triclosan.html 4. Epstein SS. The Dangers of Triclosan: A Common Antimicrobial Agent. [Online] [March 24, 2010] [Accessed April 16, 2010]. Available from: http://www.huffingtonpost.com/samuel-s-epstein/thedangers-of-triclosan_b_481323.html 5. Cookson BD. Transferable Resistance to Triclosan in MRSA. Lancet. 1991; 337: 1548 - 1549. 14 Comparison of Endospore Staining Techniques Using Bacillus cereus Wydia Davis, Brandon McElwee, Jolie Wax and Darice Wilson General Microbiology, Spring 2010 ABSTRACT Three methods were used to stain endospores of B. cereus: Fleming’s, Shaeffer and Fulton’s, and Abbott’s methods. In Fleming’s method, the background appeared black with white spaces. Abbott’s method revealed faint red outlines of vegetative cells and the Shaeffer and Fulton’s method using malachite green showed distinct green free and sub-terminal spores inside red vegetative cells. Based on this result, we conclude that the Shaeffer and Fulton’s method is the most effective method for staining endospores in a student laboratory. INTRODUCTION Endospores are formed by several pathogenic bacteria including Bacillus and Clostridium. They are of particular interest because of their ability to resist chemical and physical methods of bacterial control such as heat, ultraviolet radiation and chemical disinfectants. Due to their resistance, they can be dangerous in the field of medicine as well as in food and industrial microbiology. Endospores are impermeable to most stains and require especially harsh staining techniques such as heat. Most endospore stains are based on the concept that once they are stained, they are highly resistant to decolorization. [1] Endospore location, shape and size are consistent within a species and can aid in the identification of harmful pathogens. Until the development, in the early nineteen hundreds of some of the staining techniques for endospores, they were simply described as refractile bodies. [2] There are many methods described for the staining of endospores; however, most laboratories use malachite green as the primary stain. Several studies indicate that malachite green is toxic to mammals. It has been implicated as a multi-organ toxin which affects the functions of the liver, thyroid gland, bladder and lungs. [3] It is also said to deplete serum calcium and protein levels, while increasing the level of cholesterol. [4] As a result of this, it would be necessary to find a safer alternative to the use of malachite green in the staining of endospores, especially in student laboratories where students may not fully appreciate the issues regarding safety procedures and precautions. In this laboratory, we will compare three techniques used for the staining of endospores; Fleming’s method, Shaeffer and Fulton’s method, and Abbott’s method. Alexander Fleming’s method was introduced when India ink was no longer on the market, and therefore, employs the use of Nigrosin as a negative stain. Nigrosin is a very dark stain that does not color bacteria and therefore, provides a homologous background in which the cells and/or endospores may appear in the empty spaces. *5+ Abbott’s method uses methylene blue to color spores and anilin-fuchsin for the vegetative cells. The Shaeffer and Fulton’s method was developed in 1933 as an adaptation of Dorner’s method; it uses malachite green as the primary stain, and is the most widely used technique in laboratories today. Observations of the different techniques should reveal which is the most successful for use in the student laboratory. Our hypothesis is that the Abbott’s and Fleming’s methods are as effective as the Shaeffer and Fulton’s method for the staining of endospores. 15 MATERIALS AND METHODS Six smears of 48 to 72 hour cultures of Bacillus cereus were prepared and heat fixed onto glass microscope slides. Two smears were used for each staining technique. A large 1000 ml beaker was filled with water to the 500 ml mark and placed over Bunsen burner and heated to boil. The glass slides with the heat-fixed smears were laid flat on a staining rack on the beaker during the staining process. A strip of filter paper was placed over the smear to prevent rapid evaporation of the stain. Fleming’s method: Carbol fuchsin was applied on the smear until the entire filter paper was thoroughly soaked. The stain was left on the slide to steam for five minutes, then the slide was removed from the rack, allowed to cool, and washed with water. Thereafter, the slide was washed with 5% sodium sulfite for 8 seconds, then with water and blotted dry. A small drop of India ink was placed at one end of the slide and spread over the smear using a second slide. The stained smear was set aside to dry before examination with the oil immersion lens. Sheaffer and Fulton’s method: The smear was flooded with malachite green and left on the rack to steam for five minutes. Thereafter, the slide was removed from the rack and allowed to cool. The slide was then thoroughly rinsed with water, and counterstained with safranin for one minute. The slide was finally rinsed with water and blotted dry. The stained smear was examined with the oil immersion lens. Abbott’s method: The slide was flooded with methylene blue and allowed to steam for five minutes before washing with water. The slide was then decolorized with 95% alcohol containing 0.3 per cent HCl, and again with water. The slide was then flooded for 8 seconds with anilin-fuchsin, rinsed with water and set aside to dry. Once slides had dried, they were observed under the oil emersion lens and results were recorded. RESULTS The smears stained using the Fleming’s method showed areas of bright white against a black background. No spores could be located in the white spaces or the dark background. The smears stained with the Abbott’s method revealed pink, possibly membrane structures, with some hollow portions that did not reveal a consistent pattern to endospore location. These hollow portions had some faint bluish coloration; however, we could not determine the shape and location of the spores. The smears stained with the Shaeffer and Fulton’s method showed bright-red vegetative cells with a consistent pattern of green spores. Spores were green, located sub-terminally and were oval in shape. Free spores of the same shape and size were also identified outside of the vegetative cells. DISCUSSION Our results show that the Fleming’s and Abbott’s methods for staining spores were not reliable for identifying endospores. On the other hand, the Shaeffer and Fulton’s method was reliable for detecting the location, shape and size of the spores. This is probably responsible for its widespread use in laboratories around the world. We therefore reject the hypothesis that the Fleming’s and Abbott’s techniques are as effective as the Shaeffer and Fulton’s method for the staining of endospores. Regardless of the result of this study, laboratory instructors must pay attention to the issue of toxicity of malachite green, and therefore, caution their students to take all necessary safety precautions when 16 handling malachite green. Instructors should also insist that the staining of endospores with malachite green, should be carried out in safety hoods in order to avoid inhalation, and that students must wear protective clothing and hand gloves. REFERENCES 1. Willy J, Sherwood L. Woolverton C. Microscopes and the study of microbial structure. Prokaryotic cell structure and function. In: Prescott’s Principles of Microbiology. 1st Ed. NY, New York. McGraw Hill; 2000. p. #.[20-61] 2. Hussey M, Zayaitz A. Endospore stain protocol. 2007. [Online] [Accessed March 23, 2010]. Available from: http://www.microbelibrary.org/Edzine/details.asp?id=2565 3. Sudova E, Machova J, Svobodova Z, Vesely T. Negative effects of malachite green and possibilities of its replacement in the treatment of fish eggs and fish: a review. Veterinarni Medicina. 2007; 12: 527 – 539. 4. Srivastava SJ, Singh ND, Srivastava AK, Sinha R. Acute toxicity of malachite green and its effects on certain blood parameters of a catfish, Heteropneustes fossilis. Aquatic Toxicology. 1995; 31 (3): 241 – 247. 5. Royal College of Physicians. Sir Alexander Fleming. RCP Heritage; Munk’s Roll; Volume 5. [Online] [Accessed March 23, 2010]. Available from: http://munksroll.rcplondon.ac.uk/Biography/Details/1557 17 Bacteriological Examination of Salad: Comparing Bacterial counts Stacy-ann Wright and Katherine Foster General Microbiology, Spring 2010 ABSTRACT Microbiological contamination of vegetable salads is of great concern in the food industry. We examined vegetable salads from three food sources namely: Lincoln University cafeteria, Lincoln University Grill and Giant food store. The vegetables used were spinach (Lincoln University cafeteria and Giant food store) and lettuce (Lincoln University Grill). Representative leaves were picked from all sections of the salad container to obtain a good selection of leaves. After weighing, the salad was washed and saline dilutions of the effluent were plated on Nutrient agar. After incubation of the plates at 37 oC, the number of organisms in the neat dilutions were too numerous to count. The 1/10 dilution from the Cafeteria grew too many bacteria and could not be counted; however, 1/10 dilutions from the Grill and Giant food store were countable. For the Grill, the average count was 896 colonies/ml, and for Giant food store the average was 376 colonies/ml. Representative colonies were tested to ascertain if the bacteria were fecal in origin. None of the colonies of bacteria were positive for indole and methyl red; however, they all utilized citrate as sole source of carbon. These results indicate that the bacterial contamination of the salads were non-fecal in nature; however, the salad samples from Lincoln University had significantly more organisms than that from Giant food store. INTRODUCTION Packaged salads such as lettuce and spinach are certainly a convenience; however, there has been recent bacterial contamination in packages of pre-washed salad greens. The good news is that the contamination did not involve pathogens such as E. coli, Listeria or Salmonella; but they did find coliforms, a general indicator for bacterial infection. In the United States, there are public health standards for indicator organisms in milk, meat, drinking water, and even swimming pools; [1] however, there are no federal standards for bacteria in salad greens. Salad components can become contaminated on the farm through contact with animals, manure, contaminated water, or unsanitary handling practices during harvesting. Chlorine washes and other post-harvest treatments can help reduce surface contamination but they don’t necessarily make contaminated products safe to eat, as food-borne pathogens can be internalized in plant tissue, thereby allowing contamination to remain even after thorough washing. Even though a salad package may indicate that it has been pre-washed or triple-washed, it is still a good rule of thumb to wash these salads again. A particularly serious outbreak of deadly E. coli O157:H7 in bagged spinach in 2006 led to hundreds of illnesses, 103 hospitalizations and 3 deaths; [1] therefore, it is of importance that there are better standards and regulations throughout the salad industry. Due to the fact that there may be many organisms found in bagged salads, a good indication of contamination could be the identification of indicator organisms such as E. coli and Enterococcus. These indicator bacteria are generally not pathogenic; they live in the digestive tract of many kinds of animals. These bacteria can be used as markers of poor sanitation and bacterial 18 contamination from environmental sources like soil or from animal or human feces. Levels of these organisms are often used as an indirect measure of the potential for dangerous fecal pathogens to be present. The Food and Drug Administration, which has authority over the safety of leafy greens and all other produce items, has set no federal performance standards or bacterial limits for any indicator organisms in these commodities. Several industry experts suggest that for leafy greens, more than 10,000 colony-forming units per gram (CFU/gm) of Enterococcus, or more than 10,000 most probable number per gram (MPN/gm) of total coliforms are unacceptable levels. [1] Our hypothesis is that the samples of salad from three different food sources will yield the presence of fecal coliforms. We will therefore examine fresh salad greens from Lincoln University cafeteria, Lincoln University grill and Giant food store for bacterial contamination and identification of coliform bacteria. METHODS AND MATERIALS Fresh salad was obtained from three sources namely: Lincoln University cafeteria, Lincoln University grill and Giant food store. Leaves were picked from different parts of the salad container. Beakers and tubes were autoclaved at 121oC for 15mins. Saline solution was prepared by adding 200 ml of distilled water to 1.8 g of NaCl in a glass bottle, and mixed before autoclaving at 121 oC for 15mins. Salads were weighed in sterile beakers (sterile forceps were used to pick salad leaves). 20 ml of sterile saline solution was added to beakers containing 20 grams of salad leaves and washed thoroughly to cover all the leaves. This constituted a neat dilution from which 1/10 dilution was made. 1.0 ml of each dilution of salad effluent (neat and 1/10) was then transferred to Petri dish containing Nutrient agar. A bent glass rod was used to evenly spread the diluents on the agar. Four dilutions (two 1/10 plates and two neat plates) were plated for each salad source. The plates were allowed to dry thoroughly and were then incubated for 24 hrs at 37oC. The colonies on the agar were counted to obtain the number of colonyforming units. Representative colonies were also cultured on MacConkey broth to identify lactose fermenting and gas producing organisms; they were also Gram stained, and tested for indole production, methyl-red, Voges-Proskauer and citrate utilization. RESULTS Lincoln University Grill- Two Dilution of 1/10 (left) and two plates with no dilution (right). 19 Lincoln Univ. Cafeteria: Two Dilution of 1/10(left) and two plates with no dilution (right) Giant food Store- Two Dilution of 1/10 (left) and two plates with no dilution (right). Table 1: Plate count of bacterial colonies Plate A B Average LU Grill (1/10) 1012 780 896 Giant Food (1/10) 380 372 376 LU cafeteria (1/10) Too numerous Too numerous The results show that the colonies growing on salad from the Lincoln University cafeteria were too numerous to count even at 1/10 dilution. On the other hand, salad from Lincoln University grill yielded an average of 896 colonies per ml. Since this was a 1/10 dilution, we would have obtained a count of 8,960/ml in a neat sample of salad. The salad from the Giant Food store yielded an average count of 376/ml for a 1/10 dilution and this translates to 3,760/ml for a neat sample. Although the salad sample from Lincoln University grill fermented lactose with the production of gas in the MacConkey broth, no positive result was obtained with indole and methyl red tests. On the other hand, all samples yielded organisms that utilized citrate as the sole source of carbon. Therefore, they were non-fecal coliforms. 20 Figure 1: Fermentation of lactose with gas production DISCUSSION The salad from the Lincoln University cafeteria and Giant Food store were spinach; however, the bacterial count from the Lincoln University cafeteria were too numerous to count. Even the lettuce salad from the Lincoln University grill produced more than twice the number of colonies as the spinach from the Giant food store. This shows that irrespective of the type of salad, the bacterial contamination from Lincoln University was significantly higher than that of Giant food store. Unfortunately, due to time constraint, we could not tests salads from other stores in the area to ascertain if the situation in Giant food store was the norm. The study also showed that none of the organisms growing from the salads were of fecal origin. Our hypothesis is therefore not sustained. This is comforting because there have been several reports of salads being contaminated with fecal coliforms. We could not estimate the number of organisms from the cafeteria at Lincoln University due to the fact that the salad effluent was diluted 1/10. In future, it is recommended that a dilution of 1/100 and 1/1000 be added in order to obtain a more accurate bacterial count. This study was also carried out in the spring, so it would be necessary to repeat this study in the summer, fall and winter to determine if there are any seasonal variations in the level of bacterial contamination of vegetable salads. Finally, we intend to determine the level of contamination after every washing treatment in order to ascertain how many rinses are required to decrease the level of bacterial contamination to a minimum. In the meantime, our study indicates that there are thousands of bacterial colonies per gram of vegetables, and that salads should be thoroughly washed before consumption irrespective of what the labels say. REFERENCES 1. Bacteria and Bagged Salads: Better Standards and Enforcement Needed. Consumers Union. Yonkers, NY. February 2, 2010. [Online] [Cited 2010 April 18]. Available from: www.consumersunion.org/pdf/BaggedSaladReport.pdf 2. Wikipedia: fecal Coliforms. [Online] http://en.wikipedia.org/wiki/Fecal_coliforms [Cited 21 2010 April 18]. Available from: Comparison of Science Building with older buildings for levels of contamination of coliforms in male and female lavatories Reggie Bannerman, Stephanie F. Garcon, Ashley Lopez General Microbiology, Spring 2010 ABSTRACT We examined male and female lavatories in three buildings on the campus of Lincoln University, in order to determine the levels of coliform organisms on fomites in the facilities. We took samples from the Living Learning Center (LLC), an old student dormitory; Dickey Hall (DH), a much older academic building, and the new Science building (NSB), which was built in 2008 and has automated systems in its lavatories. We took samples from male and female lavatories in areas that were thought to be the most contaminated; the door handles (on the way out), faucet handles, soap dispensers and toilet flushers, in order to assess the cleanliness of the bathrooms. The older buildings without the automatic flushers yielded more bacteria on the surfaces of fomites. Therefore, we are recommending that renovations to the older buildings should include the modernization of the toilets to automatic flushers. INTRODUCTION Although Americans pride themselves on having good personal hygiene and clean toilet facilities, there has been little research on the cleanliness of public restrooms. [1] This experiment was designed to examine and compare the levels of contamination of coliform organisms in the lavatories of new and old buildings on the campus of Lincoln University. The older buildings have standard bathrooms, while the newer buildings include automatic systems in their bathrooms. Therefore in the older buildings, samples were collected from the toilet flushers, faucet handles, soap dispenser pumps, and the door handle (on the way out). On the other hand, in the new building, samples were only collected from the soap dispenser pumps and the door handle. Evidence exists that touch-free faucets and flushers can effectively reduce possible cross-contamination. [2] Research indicates that as many as 50 percent of all Americans do not wash their hands before leaving the washroom, thereby increasing the risk of contaminating themselves and others. Users of toilet facilities who do not wash their hands can very easily deposit potential pathogens on door knobs and these organisms can contaminate the hands of other users. People who have not used the toilets can also be affected when they shake hands with individuals who have used the toilet facilities. Microorganisms including bacteria, viruses, and parasites that have been picked up in this way can easily enter the body through hand-to-mouth or hand-to-food contact. This is of critical importance to food handlers, food service patrons, and hospital employees and patients because these germs, especially coliform bacteria, are usually indicators of fecal contamination. [1] Therefore, the microbiological examination of fomites in toilets is essential in public health. The hypothesis for this study is that the older buildings have a higher level of contamination than that of the newer building, thus providing evidence that “touch-less” bathrooms are cleaner, in terms of infectious microbes. 22 METHODS AND MATERIALS A saline solution was made by adding 0.9 grams of Sodium chloride to 100 ml of distilled water and mixing the solution until complete dissolution. Two milliliters of the saline solution was added to each of 32 test tubes. Tryptic Soy Agar was made by mixing 40 grams of Tryptic Soy Agar powder to one liter of distilled water, then, the flask containing the medium was heated until the solution boiled and became transparent. The flask containing the medium and all tubes filled with saline were then autoclaved for 15 minutes at 121°C. After cooling to 45 oC, the Tryptic Soy Agar was poured into Petri dishes and left at room temperature until solidified. To obtain samples from the toilets, a sterile cotton wool swab was dipped into a tube of sterile saline so as to wet the swab. The swab was gently pressed against the wall of the saline tube to remove any excess fluid. The wet swab was then used to swipe different surfaces of the first floor bathrooms in Dickey hall and the Science building. The swabs were then aseptically returned to the tubes of saline for transportation to the laboratory. The faucets, soap dispensers, toilet handles and door handles in the male and female bathrooms of these buildings were tested for their contamination levels. The Science building has automatic flushers and faucets, so we could only look for growth from the door handle and soap dispenser. We also took samples from the first floor men’s and women’s bathrooms in the LLC dormitory. We hypothesized that LLC would be more contaminated because a dormitory is more susceptible to bathroom use by students. After obtaining all the samples, each tube of saline containing the swab was vortexed, then, 1.0 ml of the solution was deposited on the surface of the TSA. A bent glass rod was dipped in alcohol, flamed to sterilize, cooled, and then used to spread the sample over the entire surface of the agar. The agar was incubated at 37°C, for 24 hours. The next day, each agar plate was viewed for growth of bacteria and counted. Growth from the plates was inoculated into tubes of lactose broth using a sterile loop, to see if any of the samples collected contained any lactose-fermenting bacteria that produced gas. Tubes were then incubated at 37°C for 24 hours. After the incubation period, the tubes that showed that lactose was fermented with the production of gas were smeared and Gram stained for Gram-negative bacilli. The positive tubes were also inoculated into tubes of peptone water, methyl-red and Simmons citrate and incubated at 37°C for 24 hours. After 24 hours incubation, the tubes were tested for indole production, citrate utilization, methyl red and Voges-Proskauer. For the indole test, 0.5ml of Kovac’s reagent was added to the Peptone water and shaken; a red color indicates positive while a yellow color indicates negative. For the Methyl Red test, addition of Methyl Red to the inoculated broth will yield a red color for a positive result. Lastly, for the Citrate test, the Simmons Citrate Agar, which contains bromothymol blue, will turn blue at pH of 7.6 if citrate is used as a sole carbon source. RESULTS Our results show that the male faucet handles in Dickey hall and the Living Learning Center had significantly more contamination than those of females in both buildings (Table 1). There was also more bacterial contamination of the DH male toilet flusher than that of females in the same building. However, the female faucet handles in LLC had significantly more bacteria than the female faucet 23 handles of Dickey hall. The female door in LLC also showed significantly more bacteria than that of the males or females in DH. Our results also show that samples from male faucet handles in DH and the male and female soap dispensers in the NSB fermented lactose with the production of gas. However, Gram staining of samples revealed Gram negative bacilli in DH male faucet handles and NSB male soap dispensers, and Gram positive cocci in NSB female soap dispenser. None of the samples were confirmed to be fecal coliforms because they all utilized citrate but were negative for indole and methyl red tests (Table 2). TABLE 1: Colony Count: New Science building (NSB); Dickey Hall (DH); Living Learning Center (LLC) NSB DH LLC Male Toilet Flusher NA 41 0 Male Faucet Handles NA 834 844 Male Soap Dispenser 50 12 18 Male Door 5 6 1 Female Toilet Flusher NA 2 0 Female Faucet Handles NA 2 63 Female Soap Dispenser 34 0 23 Female Door 6 5 400 NA = not applicable for Science Building because of automatic flushers TABLE 2: IMViC test results DH Male faucet Handles NSB Female Soap Dispenser NSB Male Soap Dispenser Methyl Red Negative Simmons Citrate Positive Indole Negative Negative Positive Negative Negative Positive Negative DISCUSSION Our data showed that there were no fecal coliforms identified from all the surfaces under examination; however, this was a one-time test. Therefore, there is a need for more tests on a periodic basis and at different times of the day to ascertain the true level of contamination of the toilets. In spite of the above, our results show that the male faucet handles in Dickey hall and the Living Learning Center had significantly more contamination than those of females in both buildings (Table 1). This may reflect the type of use of the toilets; whereas females use the toilets for urinating and defecating, males mostly use the toilets for defecating. In that case, the faucets may have more bacteria for the males. This also informs us that since the male faucet handles have higher levels of contamination, individuals using the facilities for whatever purpose need to wash their hands before leaving the toilets. The good news is that it appears that males using these facilities are heeding public health concerns by washing their 24 hands thoroughly, for whereas the faucet handles had very high bacterial counts, the door handles had very few bacteria. We cannot explain the rationale for the high bacterial counts in the female door handles of the LLC. It is possible that the facility has a very high number of users or that the females who use the facility are not washing their hands if they believe that they only used the facility for cosmetic purposes. Nevertheless, the females need to be informed to wash their hands thoroughly before leaving the LLC toilets. Finally, we still accept our original hypothesis that the older buildings without the automatic flushers would yield more bacteria on the surfaces of fomites. Therefore, we are recommending that renovations to the older buildings should include the modernization of the toilets to automatic flushers. REFERENCES 1. Gerber, Charles. Micro-Organisms in Public. October 1995. [Online] [Accessed March 28, 2010]. Available from: http://www.cleanseats.com/toilet_seats/view/article-7.htm 2. Lewis, Mark. Benefits of Restroom Automation: Touch-less Technology Keeps Germs in Their Place. July 2005. [Online] [Accessed March 28, 2010]. Available from: http://findarticles.com/p/articles/mi_m3830/is_7_54/ai_n14920746/ 25 Determination of bacterial contamination of public computer keyboards Jelani Johnson, Shavaughn Bowe and Michelle Haskins General Microbiology, Spring 2010 ABSTRACT Keyboards from five public computer rooms in the new Science Building were swabbed using saline solution and then, smear-plated on Nutrient Agar. After incubation at 37°C for 24 hours, discrete colonies of bacteria were significant in the Math tutoring room, Chemistry tutoring room, in left and right front areas of the main computer lab. On the other hand, the Biology and Physics tutoring rooms had minimum bacterial growth. Based on these results, we conclude that bacterial growth was significantly higher in the Math and Chemistry tutoring rooms, and the left and right front areas of the computer laboratory. This finding is consistent with the higher number of students utilizing the tutoring rooms and the computers in general computer lab. INTRODUCTION Several studies have shown that computer keyboards are reservoirs of potential pathogens. [1, 2] In the heath-care setting, keyboards have been shown to be reservoirs for pathogens because of the increased use of computers in patient care. [1] In the university setting, it has been shown that the number of organisms present in multi-user computer keyboards was significantly higher than on single user keyboards. [2] We therefore tested the keyboards in our tutoring rooms and computer lab to determine the level of bacterial contamination. The isolation of colonies can be carried out through several techniques including the spread-plate technique and pour plating. [3] In the spread-plate method, the culture has to be diluted in broth before plating occurs. This technique provides for an accurate visualization of the morphology of the bacterium. [3] It also ensures that all aerobes produce visible colonies. On the other hand, the pour-plate is very accurate as both aerobes and anaerobes are grown; however, it requires more than 48 hours for colonies at the bottom of the agar to be visualized. Also, there is the danger that fragile bacteria may be killed by heat if the medium is not sufficiently cooled before the pouring process. We therefore used the spread-plate technique to quantify the number of bacteria contaminating the keyboards. Our hypothesis is that the heavily used computers will have more bacteria on their keyboards. METHODS AND MATERIALS Tubes containing 1.0ml of saline and plates of Nutrient Agar were prepared. For the saline preparation, 0.9 grams of Sodium chloride was placed into a flask containing 100 ml of distilled water and mixed until completely dissolved. The saline solution was dispensed into tubes in 1.0ml amounts. Nutrient agar plates were prepared by suspending 23.0 grams of Nutrient agar powder in one liter of distilled water and heating to boil. Both agar and saline were then sterilized in the autoclave at 121 oC for 15 minutes. The agar was allowed to cool down to 45 oC before being poured into sterile Petri dishes. Sterile cotton-wool swabs were dipped into individual saline tubes to slightly moisten them, and the swabs were smeared over the alphabet keys only before being placed back into capped tubes. We took 26 samples from 3 computers each from the Biology, Chemistry, Physics and Mathematics tutoring rooms as well as one in three computers in the General computer room on the first floor of the Science Building. In order to determine the level of contamination, the tube containing the swab was vortexed to mix, and then, 0.1 ml of the mixture was pipetted onto agar plates in duplicates. A bent glass rod was then sterilized using alcohol and the Bunsen burner; after cooling, the bent glass rod was used to spread the mixture over the plate evenly and left to dry. After the plates had dried, they were incubated at 37°C for 24 hours. When the incubation process was complete, the bacteria was counted. RESULTS Table 1: Colony counts from keyboards in computer rooms Room Computer 1 Computer 2 Computer 3 Computer 4 Biology TR 90 20 60 Chemistry TR 2000 90 0 Math TR 10 100 1200 Physics TR 40 40 120 General CR 820 840 960 150 TR = tutoring Room; CR = computer Room Computer 5 Computer 6 120 110 The results show that the keyboards of the Biology and Physics tutoring rooms had fewer organisms ranging from a high of 120 to a low of zero, however, one keyboard each in the Mathematics tutoring room and the Chemistry tutoring room had counts of over 1200 and 2000 respectively. We also observed that while the computers in the back rows of the General computer room had less than 200 colonies on each keyboards, those in the front rows had bacterial counts in excess of 800. The front row computers are the ones mostly used by the students while the back row computers are used when those in the front are occupied. DISCUSSION The objective was not to identify the different types of bacteria on the keyboards, but rather, to quantify the number of bacteria on each keyboard. While we were not able to justify the variance in the number of bacteria in the mathematics and chemistry tutoring rooms, we were able to ascertain from the tutors that these tutoring rooms have more visitations than those of Physics and Biology. We also were able to show that the bacterial counts on the keyboards in the front rows of the general computer rooms had significantly higher number of bacteria than those at the back. In this respect, our hypothesis has been proved that computers with heavier use would be more contaminated than those with lighter use. In future, it would be useful to sample the keyboards at different times of the day and at different periods in the semester to see if there are variations in the number of bacterial counts. It would also be useful to identify the types of bacteria common to the keyboards. Irrespective of the above, it is recommended that sanitization of keyboards be adopted as a public health service in universities. 27 REFERENCES 1. Rutala WA, White MS, Gergen MF, Weber DJ. Bacterial contamination of keyboards: Efficacy and functional impact of disinfectants. Infection Control and Hospital Epidemiology. 2006; 27 (4): 372 – 377 2. Anderson G, Palombo EA. Microbial contamination of computer keyboards in a university setting. American Journal of Infection control. 2009; 37 (6): 507 – 509. 3. Willey JM, Sherwood LM, Woolverton CJ. Prescott's Principles of Microbiology. 1st ed. Boston: McGraw-Hill; 2009. 28 MICROBIOLOGICAL EVALUATION OF QUALITY OF DRINKING WATER AT LINCOLN UNIVERSITY Courtney Tipper, Marketta Williams, Laura Brown and Brian E. Cooper General Microbiology, Spring 2010 ABSTRACT Samples of drinking water from Poland Springs, Redners, Acadia, Nestle, Aquafina, Lincoln University Fountain, and Deer Park Water Cooler were tested for potability using the presumptive, confirmed, and completed test for fecal coliforms. Our results showed that only the sample from Deer Park Cooler Water contained viable bacterial growth. However, there were no fecal coliforms identified, thereby, indicating that all the water samples under test are suitable for drinking. INTRODUCTION Water is an essential compound for all living species. It permeates the vascular space of all living things; it serves as a diluent for all the chemicals and nutrients in our body fluids; we utilize water for cooking our foods and for quenching our thirst. Without water, it would be impossible for life to exist in the current form. Because of the wide use of water for everyday use, it is also an important source for the spread of infectious diseases. For example, an epidemic of cholera in London in 1840 was identified by John Snow as originating from a contaminated water source. John Snow showed that a cesspool overflow to the Thames River caused the cholera epidemic. The water and the pump used to transfer water to the community became the cause of the infection that began spreading throughout the community. Snow’s idea was to remove the pump in order to terminate the problem. [1] Since that time, the microbiological examination of drinking water has become an essential part of public health services in all developed countries. Although there are many harmless bacteria living in water, few pathogens can contaminate sources of water and result in diseases such as dysentery, typhoid, hepatitis and cholera. Testing for pathogens can be very difficult; however, the presence of Escherichia coli is an indication that the source of water is contaminated by fecal means, and therefore, unsuitable for consumption. In many homes and communities, including universities and colleges, people rely heavily on bottled water instead of the traditional tap or well water. The assumption is that these brands of water are safer for drinking than tap and well water. We therefore hypothesized that the commercially available water sources are safer for drinking than the fountain water at Lincoln University. In this study, samples of Poland Springs, Redners, Acadia, Nestle, Aquafina, LU Fountain, and Deer Park Water will be tested for the presence of fecal coliforms. MATERIALS AND METHODS The methods used for testing the water include the Presumptive Test, Plate count, Confirmed Test and Completed Test. These tests are standard tests for the examination of drinking water. For the Presumptive test, 3 double strength Lactose broth (DSLB) tubes and six single strength Lactose broth (SSLB) tubes were prepared for each of the seven samples. Each tube contained 10 ml of broth as well as a Durham’s tube for identifying gas producers. For each sample, the three DSLB were each inoculated with 10 ml of water; each of the 3 SSLB were inoculated with 1.0 ml of water, and each of 29 another set of 3 SSLB were inoculated with 0.1 ml of water. Upon adding all the samples to their respective broths, they were incubated at 35oC for 24 hours. After incubation, the tubes were observed for gas (in the Durham’s tube) and fermentation of lactose (yellow color change). All samples of water were also plated in triplicates for bacterial count. To do this, 1.0 ml of water was placed into a sterile Petri dish, and then, 20 ml of sterile, molten Tripticase Soy agar, cooled to 45oC was poured into the Petri dish. After thoroughly swirling the sample to mix, the plate was left to solidify and then incubated at 35oC for 24 hours. After incubation, plates were examined and colonies were counted. Confirmed Test All tubes of lactose broth showing the presence of acid and gas were further subjected to confirmation. The tubes were streaked unto MacConkey agar for discrete colonies which should appear pinkish for lactose fermenters. Lastly, colonies indicating the characteristics of lactose fermenters were inoculated unto single strength lactose broth and incubated at 35 oC for 24 hours for the production of acid and gas. Completed Test All colonies appearing pinkish on MacConkey agar were Gram-stained for the presence of Gramnegative bacilli. The single strength broths were also examined for the production of acid and gas. RESULTS Results from the presumptive test showed that only the water sample from Deer Park cooler water was positive for gas and acid in the lactose broth. The other water samples were negative for the presumptive test and did not produce any viable colonies on the agar plate. No further test for confirmation was therefore required for these water samples. Further confirmation of Deer Park cooler water (DPCW) sample, revealed the presence of Gram-positive bacilli, and none of the samples showed a positive indole or methyl red test. Therefore, they were not fecal coliforms. Table 1: Presumptive test results in lactose broth DPCW LUWF Nestle Redner’s Arcadia Aquafina Poland Spring 0.1 ml Gas Fermentation No Growth No Growth No Growth No Growth No Growth No Growth 1.0 ml Gas Fermentation No Growth No Growth No Growth No Growth No Growth No Growth 10 ml Gas Fermentation No Growth No Growth No Growth No Growth No Growth No Growth Concentration 30 Table 2: Water samples spread over TSA Gram Negative Stain Colonies Shape Plate 1 Plate 2 Plate 1 Plate 2 Plate 1 Plate 2 DPCW Gram Positive Gram Negatve Some in clusters, some spread out Clusters spread out Bacilli (rods) Long bacilli (rods) LUWF No Growth No Growth No Growth No Growth No Growth No Growth Nestle No Growth No Growth No Growth No Growth No Growth No Growth Rednerrs No Growth No Growth No Growth No Growth No Growth No Growth Arcadia No Growth No Growth No Growth No Growth No Growth No Growth Aquafina No Growth No Growth No Growth No Growth No Growth No Growth Poland Spring No Growth No Growth No Growth No Growth No Growth No Growth Table 3: Cross contamination check of positive water sample DPCW Gram Negative Stain Colonies Shape Plate 3 Plate 3 Plate 3 Gram Positive Clusters Short bacilli (rods) DISCUSSION This study showed that all the water samples contained no fecal coliforms and therefore, suitable for drinking. Only the Deer Park cooler water sample contained any viable bacteria, and showed positive 31 presumptive test. The fact that the Lincoln University water fountain was as clean as the other samples, and in fact better than the Deer Park cooler water, proves that our hypothesis that commercially available water is better than the traditional water source is not acceptable. Therefore, students and the administration need not spend scarce resources to pay for water of the same standard as traditional water sources. Also, the use of bottled water generates a lot of plastic containers that need to be disposed as they are not biodegradable. Therefore, in order to preserve the environment, the drinking of water from traditional sources should be encouraged as long as these sources are regularly tested to ensure potability. LITERATURE CITED 1. Bacteriological Examination: Qualitative Tests. Microbiology Laboratory: theory and application. Brief edition, Englewood, Colorado. Morton Publishing Company; 2008: p. 359-362 32 The Effectiveness of Alcohol based and Non- Alcohol based Mouthwashes against Staphylococcus aureus and Pseudomonas aeruginosa S. Bennett, K. Baugh, N. Pollard, Jaleesa Johnson, R. Madlock, O. Anyanwu General Microbiology, Course 2010 ABSTRACT The objective of this experiment was to evaluate the effectiveness of alcohol-based and non-alcohol based mouthwashes in killing Staphylococcus aureus and Pseudomonas aeruginosa. Dilutions of alcoholbased and non-alcohol based mouthwashes were tested on S. aureus and P. aeruginosa for 30 seconds. Our studies showed that non-alcohol-based mouthwashes were more effective in killing the test organisms than alcohol-based mouthwashes. We can therefore conclude that the addition of alcohol in mouthwashes is not effective in killing bacteria and should be discontinued. INTRODUCTION The human mouth contains millions of bacteria, most of which are not pathogenic. However, the bacteria that naturally exist in the mouth can build up into a plaque if good oral hygiene is not practiced. Mouthwashes are most commonly used to fight cavities, remove bad breath and prevent plaque buildup. There are two main forms of mouthwashes, alcohol-based and non-alcohol based. Alcohol destroys bacteria by first extracting lipids from the outer protective cell membrane, thereby increasing solubility in water, [1] and facilitating the disintegration of cell membranes. After disintegration, alcohol can then enter the cell and modify proteins within each bacterium. Alcohol is one of main ingredients in most mouthwashes and serves as a solvent, antiseptic, and preservative to active ingredients. Mouthwashes are liquid preparations intended for application on teeth and mucosa of the oral cavity and pharynx in order to exert antiseptic, astringent and sedative local action. [2] It is also added to allow the diffusion of essential ingredients such as menthol, eucalyptol and thymol into plaques. In spite of the above justification for the addition of alcohol in mouthwashes, several research studies have reported finding an association between long-term alcoholic mouthwash use and oral cancer. [3, 4, 5] For example, in a study conducted by Wynder and colleagues, [3] they found a significant association between alcohol mouthwash use and oral cancer. A bigger multi-site study by Guha and colleagues, [5] comparing participants who reported having used alcohol mouthwash to those who reported never having used mouthwash, found that individuals who reported using mouthwash more than twice a day were nearly six times more likely to develop oral, squamous cell carcinoma, compared to those who reported never having used mouthwash. Oral cancer is diagnosed in 5,000 people with 1,600 deaths each year in the United States. [4] Alcohols also cause burning, dryness and mouth pain in some users. It is therefore essential to determine if there is an enhanced benefit in the quality of mouthwashes from the addition of alcohols. If there are no perceived benefits, there would be no need to add an ingredient that could cause diseases like oral cancers. This experiment is designed to compare the effectiveness of mouthwashes containing alcohol versus mouthwashes, which do not contain alcohol in killing Staphylococcus aureus (Gram positive cocci) and Pseudomonas aeruginosa (Gram negative bacilli). Our 33 hypothesis is that alcohol-based mouthwashes have greater antibacterial property than non-alcoholbased mouthwashes. MATERIALS AND METHODS 150 ml of Tryptic Soy Broth was made and 5 ml of the broth was placed in each of 24 tubes. The 24 tubes containing the broth along with 24 empty tubes and a flask of distilled water were sterilized by autoclaving at 121oC for 15 minutes. Four different mouthwashes were used: two alcohol based (Green Mint mouthwash and Natural Citrus mouthwash) and two non-alcohol based (Green Mint mouthwash and Blue Crest mouthwash). Two sets of serial doubling dilutions of each mouthwash was done in the sterile tubes using distilled water as diluent to obtain dilutions of 1/2; 1/4; 1/8; 1/16. A control tube containing saline without mouthwash was included in the test. Overnight broth cultures of Staphylococcus aureus and Pseudomonas aeruginosa were diluted in saline to McFarland 0.5 and used for the test. 50 µl of the test organisms was added to each dilution of the mouthwash and the saline control and vortexed. After thirty seconds (the recommended swishing time for mouthwashes), 50 µl of the contents of each tube was removed and inoculated into a tube of sterile Tryptic Soy Broth. The tubes of broth were mixed and incubated at 37oC for 24 hours. After the incubation, the broth was examined for no growth, indicating the killing of the bacteria or growth, indicating the survival of the bacteria. RESULTS Table 1: Effect of Alcoholic Based Mouthwash on bacteria Alcohol Based: Staphlococcus auerus Pseudomonas aeruginosa No bacteria Alcohol Based: Natural Citrus Mouthwash Neat Green Mint Mouthwash Neat 1/2 No bacteria 1/2 No bacteria 1/4 Bacterial Growth 1/4 Bacterial Growth 1/8 Bacterial Growth 1/8 Bacterial Growth 1/16 Bacterial Growth 1/16 Bacterial Growth Saline control Bacterial Growth Saline control Bacterial Growth No bacteria The results of our study as shown in Table 1 indicate that the two brands of alcohol-based mouthwash killed the test bacteria in 30 seconds at undiluted and at 1/2 dilution; however, no test bacteria were killed when the mouthwash was diluted from 1/4 to 1/16. There was no difference in the killing of both bacteria by both brands of alcohol-based mouthwash irrespective of the fact that S. aureus is a Gram positive coccus and Pseudomonas aeruginosa is a Gram-negative bacillus. 34 Table 2: Effect of Non- Alcoholic Based Mouthwash on Bacteria Non- Alcoholic Based: Green Mint Mouthwash Staphlococcus auerus Non- Alcoholic Based: Blue Crest Pseudomonas aeruginosa Neat No bacteria Neat No bacteria 1/2 No bacteria 1/2 No bacteria 1/4 No bacteria 1/4 No bacteria 1/8 No bacteria 1/8 No bacteria 1/16 No bacteria 1/16 No bacteria Saline Bacterial Growth Saline Bacterial Growth Table 2 indicates that the two brands of non-alcohol-based mouthwash killed the test bacteria at dilutions of neat to 1/16. Both types of bacteria were equally killed. The fact that the control tube showed the presence of growth indicates that the bacteria were alive and the test procedure was accurate. DISCUSSION In this experiment, we were able to compare the effectiveness of two brands of alcohol-based mouthwashes and non-alcohol based mouthwashes against Staphylococcus aureus and Psuedomonas aeruginosa. Based on these results, we discovered that more bacteria grew in the Alcohol BasedNatural Citrus with the Pseudomonas aeruginosa bacteria and the Alcohol Based-Green Mint Mouthwash with the Staphlococcus auerus except for the neat (pure mouth wash with no dilution) and the 1/2 dilution of mouthwash. However, no bacteria were found in the test tubes containing nonalcohol-based Green Mint with Staphylococcus aureus, non-alcohol-based-Green Mint and non-alcohol based-Blue Crest with Pseudomonas aeruginosa except for the saline which was used as a control for the experiment. Therefore, it was proved that Non-Alcohol Based mouthwash is more effective in killing bacteria than Alcohol Based mouthwash. Non-Alcohol Based mouthwash can certainly clean and disinfect our mouths, teeth, gums, and breath without the necessity of alcohol as a solvent. The result of this study therefore does not support our hypothesis that alcohol-based mouthwashes are more effective in killing bacteria than non-alcohol-based mouthwashes. Given the problems of increased risk of oral cancers, mouth dryness and sensitivity from using alcohol-based mouthwashes, the results of this study prompts us to recommend that alcohol should not be used in mouthwashes. 35 REFERENCES 1. Samantha Herman. eHow. How Does Mouthwash Kill Bacteria? [homepage on the Internet]. 2009 [cited 2010 Apr 15]. Available from: http://www.ehow.com/how-does_4899881_mouthwash-killbacteria.html 2. Carretero-peláez, Esparza-gómez, Figuero-ruiz E. Alcohol-containing mouthwashes and oral cancer. Critical analysis of literature. In Medicina Oral, Patología Oral Cirugía Bucal (Ed. impresa). Valencia: Medicina Oral S.L; 2004 3. Weaver A, Fleming SM, Smith DB. Mouthwash and oral cancer: carcinogen or coincidence? Journal of Oral Surgery. 1979; 37:250-253. 4. Wynder E L, Kabat G, Rosenberg S, Levenstein M. Oral cancer and mouthwash use. J National Cancer Institute. 1983; 70: 255-260. 5. Guha N, Boffetta P, Wunsch Filho V et al. Oral health and risk of squamous cell carcinoma of the head and neck and oesophagus: results of two multicentric case-control studies. American Journal of Epidemiology. 2007; 166: 1159-1173. 36 Section 2: Research papers 37 Syphillis Kaylene Baugh General Microbiology, Spring 2010 INTRODUCTION Syphilis is a sexually transmitted disease caused by a spirochete, Treponema pallidum. In the primary stage of infection, symptoms include painless sores and swollen lymph nodes. Those with secondary syphilis may also have fever, fatigue, rash, aches and pains, and loss of appetite, among other symptoms. Tertiary syphilis causes heart, brain, and nervous system problems. Because of the complications and severity of tertiary syphilis, it is important that patients receive treatment in the primary and secondary stages of the illness. Syphilis is also important as a research paper because it is involved in one of the best described experiments with human subjects in Tuskegee. This study which used African Americans in the study of a disease, is one of the reasons that experiments involving human subjects are rigorously controlled and need the informed consent of the participants. This research paper presents the general description of the disease, the significance, the general symptoms, pathogenesis, laboratory diagnosis, treatment and methods of prevention. GENERAL DESCRIPTION OF THE DISEASE Syphilis is a complex sexually transmitted disease (STD) caused by the spirochetal, Gram negative bacterium named Treponema palilidium. Treponemes have a helical shape and a double membrane. The bacteria move by means of axial filaments (internal flagella) and use carbohydrates as their only source of energy. Treponema cannot synthesize fatty acids, enzyme cofactors, and most amino acids, and are dependent on their host for their acquisition. They are unable to grow in-vitro in artificial medium due to the bacterium's complex, and as yet, unknown, nutritional requirements. Treponema does not have surface proteins and as a result of this absence, it has been impossible to create vaccines. *1+ Syphilis is often called “the great imitator” due to the fact that its signs and symptoms are indistinguishable from those of other diseases especially when it is in its late stages. This disease is passed from one person to the next through direct contact with a syphilis sore. These sores are mainly found on the external genitals, vagina, anus or in the rectum. Sores can also be found in the mouth or on the lips. The organism is transmitted during vaginal, anal or oral sex. The disease can also be passed from infected, pregnant mothers to their unborn children. Syphilis cannot be transmitted through physical contact. Syphilis is considered to have different stages; primary, secondary, latent and tertiary syphilis. If left untreated, this disease can damage the heart, aorta, brain, eyes and bones. In some cases, these effects can also lead to death. It is known that the genital sores also make it easier to transmit and acquire HIV and other sexually transmitted infections. THE SIGNIFICANCE OF SYPHILIS Syphilis is a genital ulcerative disease that causes a substantial amount of complications if left untreated, and can also facilitate the transmission of HIV and other sexually transmitted diseases. 12 million cases occur each year worldwide; in the US, there are about 100,000 cases annually. [1] Congenital syphilis 38 affects about 8.5 per 100,000 live births in the US and is on the increase. The prevalence is about 4 per 100,000 in the US. [1] Up to 40% of syphilis cases involving pregnant women result in perinatal death. It has been found that in 80% of cases, the pregnant mother acquires the disease up to four years prior to the pregnancy. [1] This is due to the asymptomatic nature of the disease between the primary and secondary stages of infection. However, the rates of primary and secondary syphilis have been decreasing over the years since 1990s. In the United States primary and secondary syphilis declined 89.7% between 1990 and 2000, although the rates have increased annually between 2001 and 2008. [2] Overall, increases in rates between 2001 and 2008 were observed primarily among men (increasing from 3.0 cases per 100,000 population to 7.6 cases per 100,000 population). After several years, 40% of untreated people in the secondary phase enter a tertiary phase. This phase is characterized by degenerative lesions (gummas) in the skin, bone and nervous system due to hypersensitivity reactions. [1] Increases in cases among MSM (including men who have sex with women and men) have occurred and have been characterized by high rates of HIV co-infection and high-risk sexual behaviors. There has been an increase from 4% in 2000 to 62% in 2004 for the estimated rate of primary and secondary syphilis cases attributable to MSM. 63% of the primary and secondary syphilis cases in 2008, found in 44 states and Washington, DC were among MSM. [1] Treatment in the primary and secondary stages is effected with penicillin. In later stages, the disease is harder to treat. In 2008, the rate of primary and secondary syphilis was highest in persons in the 20 to 24-year-old and 25 to 29-year-old age groups (11.4 and 10.7 cases per 100,000 population, respectively). Syphilis reflects one of the most glaring examples of racial disparity in health status, with the rate of African Americans nearly 30 times the rate for Caucasians. [3] GENERAL SYMPTOMS OF SYPHILIS Syphilis can be characterized into four stages namely the primary, secondary, latent and tertiary. When a person becomes infected with syphilis, there is a period of 9 to 90 days called the incubation period (the average being around 21 days) before the first signs and symptoms of the disease appear. For every stage of the disease, there are characteristic signs and symptoms, but any particular sign or symptom may or may not be present. The primary stage of syphilis is usually identified with a sore called a “chancre” on the skin that is initially exposed to the infection, and is usually found in the genitals, rectum or mouth. The chancre is usually described to be firm, round, small and painless and measuring half-an-inch across. A painless swelling of the lymph nodes of the groin may also occur. The chancre heals spontaneously after 4 to 6 weeks and infected individuals generally do not feel ill in the primary stage of syphilis. If adequate treatment is not administered, the infection progresses to the secondary stage. Secondary syphilis can often occur several weeks after the chancre heals, once the bacteria have spread through the body. When the disease reaches the secondary stage, an individual may feel sick and the symptoms include achiness, headache, loss of appetite and maybe rash. The rash in secondary syphilis is usually reddish-brown in color, not itchy and widespread. However, the appearance of the rash's individual lesions can vary dramatically: they may be flat or raised; they may or may not be scaly; and 39 there may or may not be pustules present. One of the reasons why syphilis is deemed “the great imitator” is because of this rash and due to the fact that it can resemble many other conditions. The rash can last up to months. The secondary stage of syphilis includes other symptoms such as sore in the mouth, nose, throat, and on the genitals or folds of the skin. It is common for victims to have swelling of the lymph nodes and patchy hair loss. The symptoms and signs of the secondary stage of syphilis will disappear without treatment in 3 weeks to 9 months, but the infection will still be present in the body. The latent stage of syphilis occurs after the symptoms of the secondary stage have disappeared, and this stage can last up to 50 years. The latent stage has no symptoms, and an infected person may cease to be contagious after 2 years. Nonetheless, a person in this stage is still infected and the disease can be diagnosed by a blood test. An infected pregnant woman can transmit syphilis to her fetus during the latent stage. The final stage of syphilis known as the tertiary stage occurs in about one third of those who are not treated. During the latent stage the disease remains in the body and it may begin to damage the internal organs including the brain, nerves, eyes, blood vessels, heart, bones, joints and liver. Common symptoms of this stage may include fever, painful non-healing skin ulcer, bone pain, liver disease and anemia. Syphilis in the tertiary stage can also affect the nervous systems and the aorta, resulting in the loss of mental function and heart disease respectively. PATHOGENESIS The pathogenesis of a disease provides clear understanding about the disease; however, the mechanisms by which host–pathogen interactions influence the course of syphilis have been compromised by the fact that the organism cannot be grown in vitro and, as an exclusively human pathogen, inferences made from animal models are of limited applicability. [4] Humans are the only natural host for T pallidum subsp pallidum, and infection occurs through sexual contact. The organisms penetrate mucous membranes or enter minuscule breaks in the skin. Experiments in both humans in Tuskegee and rabbits using standardized inocula indicate that less than 10 organisms are capable of producing infection. In women, the initial lesion is usually on the labia, the walls of the vagina, or the cervix; in men, it is on the shaft or glans of the penis. A chancre also may occur on the lips, tongue, tonsils, anus, or other skin areas. [4] The observation, made in a number of in vitro studies, that T pallidum subsp pallidum and subsp pertenue specifically attach to numerous cell types is believed to reflect the ability of these bacteria to infect diverse tissues and organs. Despite a lack of neurological symptoms, treponemes often can be recovered by rabbit inoculation with either normal or abnormal spinal fluids, and a high proportion of patients with early syphilis have CSF abnormalities. [4] The genome of T. pallidum was sequenced in 1998. The organism has a relatively small genome, suggesting that it utilizes host biosynthesis to fulfill some of its metabolic needs. The Tp genes, identified as a family of repeat genes, encode proteins homologous to the major sheath proteins of T. denticola. [4] Through gene conversion, antigenic variation has been hypothesized to be one mechanism of escaping immune surveillance, prolonged infection, and persistence in the presence of a robust host response. Intense inflammatory responses and prostaglandins, induced by fetal infection during 40 pregnancy, may be responsible for fetal death or pre-term delivery and severe growth retardation or other manifestations of congenital syphilis. [2] Syphilis is a chronic, bacterial and sexually transmitted infection that is characterized by the primary chancre and disseminated secondary rash, lesions that initially contain large numbers of Treponema palilidium but resolve spontaneously after several weeks. Bacterial clearance and lesion resolution in early syphilis is aided by the phagocytosis of Treponema pallidum. The fact that secondary syphilis lesions containing high numbers of bacteria develop in the presence of high antibody titers against treponemal antigen demonstrates the inability of humoral immunity to control infection. It has been shown in histologic studies that major infiltrating cells in syphilis are CD4 and CD8 T-lymphocytes and macrophages. The number of treponemes drastically decreases shortly after macrophage infiltration. [4] The treponemal antigen can then be seen within the healing of the macrophage lesion. The fact that macrophages can ingest and kill T. palilidium in-vitro is an indication that macrophages are responsible for the clearance of bacteria from these infected tissues. Although the importance of the cell-mediated immunity in syphilis has been recognized, there is no information about the cytokines present in syphilis lesion that initiate the local immune response. [5] LABORATORY DIAGNOSIS Clinical testing is a very important part of the diagnosis of syphilis due to the numerous clinical manifestations of the disease. Treponema pallidum, the etiological agent cannot be cultured, and there is no single optimal alternative test. The most frequently used approach in laboratory diagnosis of syphilis is the serological testing. There are various serological and alternative tests currently available. There is a great need to use multiple tests and to include quality control. Due to the complexity of syphilis, the services of reference laboratories and clinical experts are often needed. Due to the diverse clinical manifestations that syphilis possesses, it shares many clinical features with other treponemal and non-treponemal diseases. Therefore, it is mandatory that the clinical diagnosis is always supported by appropriate laboratory tests and that the test results are interpreted with reference to the patient's history and physical examination findings. Syphilis progression occurs through distinct primary, secondary, latent and tertiary stages. The ulcers that appear in primary and secondary syphilis are rich in treponemes; venereal transmission occurs through direct contact with these lesions. *6+ The patient’s presenting symptoms have implications for diagnosis and treatment at this stage of the disease. The disease may be asymptomatic in some stages, and there are problems in diagnosing very early syphilis, asymptomatic congenital syphilis, neurosyphilis and syphilis in intravenous drug users and persons co-infected with serologically cross-reacting agents and HIV. There are many tests for the direct and indirect diagnosis of syphilis, although T pallidum cannot be grown in culture. [6] Microscopic examination of fluid or smears from lesions, histological examination of tissues or nucleic acid amplification methods such as polymerase chain reaction (PCR) are direct diagnostic methods that include the detection of T pallidum. Indirect diagnosis is done using serological tests for the detection of antibodies. [6] 41 There are two categories of serological tests; non-treponemal tests for screening, and treponemal tests for confirmation. Both immunoglobulin IgG and IgM, antiphospholipid antibodies formed by the host in response to lipoidal material released by damaged host cells early in infection and lipid from the cell surfaces of the treponeme itself are measured in all non-treponemal tests. On the other hand, T pallidum or its components are the antigens used for all treponemal tests. [6] Latent syphilis can only be diagnosed by serological tests. There are no reliable routine laboratory methods known to distinguish the other pathogenic treponemes from each other or from syphilis. Direct diagnostic tests include detection of T. pallidum by microscopic examination of fluid or smears from lesions, histological examination of tissues or polymerase chain reaction (PCR). Specimens include exudates from lesions of primary, secondary and early congenital syphilis. Exudates and fluids are examined as a wet mount using dark-field microscopy. Identification is based on the characteristic morphology and motility of the spirochete. Dark-field microscopy requires trained, experienced microscopists. Also, treatment with antibiotics may result in a false-negative finding. Direct fluorescent antibody test is easier to perform than dark-field microscopy. It detects treponemal antigens and does not require motile treponemes. The test uses fluorescein-labeled antibody specific to pathogenic treponemes, and is suitable for specimens from oral and rectal lesions. Direct fluorescent antibody tests can also be used to detect T. pallidum in tissue sections. The last direct test involves the use of PCR which can detect as low as one to ten organisms per specimen. The PCR method is useful for monitoring treatment, and can differentiate between old and new infections. Non-treponemal tests involve the detection of antibodies. They become positive one to four weeks after the appearance of the primary chancre, and six weeks after exposure. The tests use antigen containing standardized amounts of cardiolipin, cholesterol and lecithin. Tests approved by the CDC include: Venereal Disease Research Laboratory (VDRL); Rapid Plasma Reagin (RPR); Unheated Serum Reagin (USR); and Toluidine Red Unheated Serum Test (TRUST). Non-treponemal tests are rapid, simple and inexpensive; they can also detect re-infection. The main limitations include reduced sensitivity in primary and late syphilis, false positive results due to cross-reactivity, and potential for false negative due to prozone reactions. TREATMENT OF SYPHILIS Unlike several other diseases, there is a cure for syphilis. Effective treatments for this disease are antibiotics. The most widely used antibiotic is penicillin. The dose of the drug and how it is given is dependent on the stage of the disease. For those individuals who are allergic to penicillin, there are alternative drugs called doxycycline. [7] A reaction called Jarish-Herxheimer may occur several hours after treatment of the early stages of syphilis. The symptoms of this reaction may include chills, fever, general feeling of being ill, general joint aches, general muscle aches, headache, nausea and rash. The symptoms; however, usually disappear after 24 hours. Follow-ups for blood test should occur at 3, 6, 12, and 24 months to ensure that the infection has been eliminated. [7] The infected individual should abstain from sexual activities until two follow-up tests show that the infection has been cured. Treatment will kill the syphilis bacterium and prevent further damage, but it will not repair damage already done. 42 PREVENTION Abstinence from sexual activities or engaging in a long-term mutually monogamous relationship with a partner who has been tested, and is known to be uninfected, is the surest ways to avoid the transmission of sexually transmitted diseases, including syphilis. There are other activities such as engaging in alcohol drinking and drug-use that could contribute or lead to risky sexual behaviors that could result in the contraction of syphilis or other sexually transmitted diseases. It is also very important that sex partners communicate effectively so they can discuss their status and history on STD’s so that preventative actions can be taken. [1] Correct and consistent use of latex condoms can reduce the risk of syphilis, as well as genital herpes and chancroid, only when the infected area or site of potential exposure is protected. [7] It should be known that condoms lubricated with spermicides (especially Nonoxynol-9 or N-9) are no more effective than other lubricated condoms in protecting against the transmission of STDs. Use of condoms lubricated with N-9 is not recommended for STD/HIV prevention. Syphilis and other STDs cannot be prevented from transmission by washing the genitals, urinating, and/or douching after sex. One should refrain from sexual activities and see a physician immediately if any unusual discharge, sore, or rash, particularly in the groin area is seen. [7] Prevention strategies also include testing blood before transfusion to ensure that T. pallidum is not transmitted during transfusion. Pregnant women can avoid infection to their children by taking blood tests, and receiving treatment if they are infected. REFERENCES 1. John Chikwem. Chlamydia - Description PowerPoint Presentation. [Online] [Cited 4 April 2010]. Available from: WebCT: http://webct41.lincoln.edu:8900/SCRIPT/092SBIO40101/scripts/serve_home 2. The laboratory diagnosis of syphilis. National Center for Biotechnology Information. [Online] [Cited 17 March 2010] Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2095002 3. The American Journal of Nursing: Syphilis- Health disparities, prevalence and incidence of this disease. Volume 38, No. 2. [Online] [Cited 16 March 2010]. Available from: http://www.uchicago.edu/journal/jstor 4. JSTOR: Syphilis [Online] [Cited March 17, 2010] http://www.jstor.org/stable/3413777?&Search=yes&term=syphilis&list Available from: URL: 5. Chicago Journals - The Journal of Infectious Diseases." [Online] [Cited 22 Apr. 2010]. Available from: URL: <http://www.journals.uchicago.edu/doi/abs/10.1086/511822>. 6. Byrne RE, Laska S, Bell M, Larson D, Phillips J, Todd J. Evaluation of Treponema pallidum. "The laboratory diagnosis of syphilis." National Center for Biotechnology Information. [Cited 22 Apr. 2010]. Available from: URL: <http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2095002/>. 7. STD Facts – Syphilis: Centers for Disease Control and Prevention. [Online] [Cited 16 March 2010] Available from: URL: http://www.cdc.gov/std/syphilis/STDFact-Syphilis.htm#WhatIs 43 BOVINE SPONGIFORM ENCEPHALOPATHY Brian E. Cooper General Microbiology, Spring 2010 GENERAL DESCRIPTION OF DISEASE Bovine Spongiform Encephalopathy (BSE), also known as mad cow disease, is a degenerative neurological disorder of cattle. This fatal disease that affects the central nervous system (CNS) is caused by a transmissible agent known as a prion. [1] BSE is one of many types of diseases called transmissible spongiform encephalopathies (TSEs). TSEs include scrapie, which affects sheep and goats; transmissible mink encephalopathy; chronic wasting disease of deer and elk; and in humans, kuru, both classic and variant Creutzfeldt–Jakob disease (CJD), Gerstmann–Straussle–Scheinker syndrome, and fatal familial insomnia. [2] In 1986, it was first diagnosed in the United Kingdom, and has been responsible for the death of numerous cattle since then. SIGNIFICANCE As a disease, BSE is important for many reasons. The first cases of BSE were found in cows during the 1970's with two cases of BSE being identified in 1986. BSE possibly originated as a result of feeding cattle meat and bone-meal that contained BSE-infected products from a spontaneously occurring case of BSE or scrapie-infected sheep products. [1] Scrapie is a neurodegenerative disease in sheep caused by prions. The outbreak was amplified and spread throughout the United Kingdom cattle industry by feeding rendered, prion-infected, bovine meat and bone-meal to young calves. [1] Cases of the epidemic have been found in many European countries along with countries outside of Europe, including Japan, Canada, and the United States. Based on World Organization of Animal Health (OIE) standards for BSE surveillance, the reported national prevalence rates of BSE in North American cattle, particularly in animals born in the United States, is very low, and therefore, difficult to measure accurately. [1] However, more than 95 percent of the total worldwide cases have occurred in the United Kingdom. [2] The epidemic in the United Kingdom peaked in January 1993 at almost 1,000 new cases per week. As of the end of 2008, there were over 184,500 confirmed cases of BSE in the United Kingdom alone. This number included more than 35,000 herds. [1] Due to the large number of cases and the egregious nature of the problem, many cattle were euthanized along with the depletion of cattle use in the food industry for both humans and animals. For example, in 1989, over 8000 suspected and confirmed cases of BSE were slaughtered. About 70 percent of the slaughtered animals were disposed of by incineration and the rest by burial at approved sites. The compensation costs for the year were over £2.8 million and the disposal costs amounted to £1.6 million. [3] In the winter of 1989 and 1990, the United Kingdom Government banned the human consumption of foods made with certain bovine offal (entrails and internal organs) that were associated with large numbers of BSE infection. The same specified offal was banned from use in feed for all mammals and birds. Many measures such as the ones above were implemented. Though, these same measures had 44 no impact in preventing several countries from banning imports of a much wider range of human and animal food products containing bovine tissues other than the proscribed offal. This has seriously disrupted the United Kingdom export trade. [3] Besides having a fairly large economic impact in the U.K., the compensation for loss of products due to the ceasing of trade also spurred changes in the economic markets of those countries that received decreased imports of banned products. It can also be assumed that the consumer trust in bovine products dwindled as a result of BSE’s presence. GENERAL SYMPTOMS The incubation period of BSE averages 4 to 6 years, although the period can be longer or shorter. [2] However, upon the onset of clinical signs, the disease becomes fatal or causes crimpling destruction of the CNS. This process of deterioration usually takes from 2 weeks to 6 months. [2] Most infected cattle were likely exposed to the agent as calves and became infected during the first year of life. This suggests that susceptibility in cattle declines with age; therefore, young animals are most susceptible. [2] The incubation period is inversely related to dose. Therefore, an animal exposed to a small amount of the BSE agent would have a longer incubation period than an animal exposed to a large amount of the agent. [2] In cattle, BSE does not cause any detectable immune response or inflammatory reaction in host animals. [2] The symptoms from the progressive degeneration of the nervous system include changes in temperament, such as nervousness or aggression; abnormal posture; lack of coordination and difficulty in rising; decreased milk production; or loss of body weight despite continued appetite. [4] Other symptoms include apprehension, fear, increased startle, or depression; hyper-aesthesia or hyperreflexia; adventitial movement (muscle fasciculations, tremor and myoclonus); ataxia of gait, including hypermetria; and autonomic dysfunction (reduced rumination, bradycardia and altered heart rhythm). [5] BSE also affects the brain and spinal cord of cattle. Lesions are characterized by sponge-like changes visible with an ordinary microscope. [6] It has been observed that sometimes symptoms may be slight, undetectable or unrecognizable and that not all cattle with BSE show all symptoms of the disease. [4] PATHOGENESIS The pathogenesis of BSE is not clearly known. One theory proposes that the agent is made of a selfreplicating protein called prion. Another theory suggests that the agent is virus-like and possesses nucleic acids which carry genetic information. Although, there is strong evidence that supports the cause being prions, the ability of the BSE agent to form multiple strains is more easily explained by a virus-like agent. [6] The normal prion protein changes into a pathogenic form that damages the central nervous system of cattle. [1] The transmission of BSE is understood. BSE is not a contagious disease and it is not transmitted through direct contact between animal and animal nor animal and human. The primary means by which animals become infected is through consumption of feed contaminated with the infectious BSE agent, prions. [7] Research shows that the contamination of feed stems from adding ingredients that contain protein from rendered infected cattle. Since the rendering processes does not completely disable or kill the BSE agent, rendered protein such as meat and bone-meal derived from infected animals may contain the 45 infectious agent. Infectivity has only been found in brain tissue, the spinal cord, as well as the retina of the eye in naturally infected cattle. On the other hand, in experimentally infected cattle, the distal ileum of the small intestine, tonsil, dorsal root ganglion, and trigeminal ganglion also were found to be infective. [2] This research involved inoculating tissues from infected cattle into mice to determine if disease transmission occurred. This process of inoculation was also repeated with cattle instead of mice. [2] Due to limited research, maternal and vertical transmission cannot be ruled out. Scientific evidence suggests that this is unlikely to occur significantly, if at all. Also, maternal transmission only accounts for about 10 percent of the offspring of all BSE cases. Furthermore, recent epidemiological research indicates that maternal transmission likely does not exist. [4] There is no evidence supporting the transmission of BSE through milk. [4] Finally, epidemiological evidence suggests the epidemic of this disease has passed its peak. [4] LABORATORY DIAGNOSIS No test currently exists to detect the disease in live cattle. However, veterinary pathologists confirm BSE by microscopically examining the brain tissue or by the detection of abnormal prions in brain tissue. [2] Hence BSE is named due to the spongy appearance of the brain tissue of infected cattle when examined under a microscope. Other testing methods include immunohistochemistry of the BSE fibrils. Unlike the previous tests whose results take up to a week, more rapid tests that provide results within 36 to 48 hours have been developed to detect the abnormal prion in brain or spinal cord tissue of dead animals. These rapid tests can be used to determine if BSE exists in a population and to obtain an indication of its prevalence, or to detect animals with the disease that have yet to show clinical signs. [7] TREATMENT Unfortunately, there is no existing treatment to fight against or prevent BSE. Those cattle that become infected eventually die naturally or are killed. The agent is highly stable, resisting freezing, drying and heating at normal cooking temperatures, even those used for pasteurization and sterilization. [6] PREVENTION The most effective means of prevention is eliminating the use of mammalian proteins for feeding ruminants, more specifically cattle. This ban, announced by the FDA in 1997, was one of the most important measures in preventing the transmission of the BSE disease to cattle. Animal and Plant Health Inspection Service (APHIS), has taken aggressive measures to prevent the introduction and potential spread of BSE in the United States. APHIS has maintained stringent restrictions since 1989 to prevent importation of the highest risk animals and products. [2] One of the most important public health protective measures is the removal of high risk or infectious materials from the human food supply. Also, other controls include banning non-ambulatory, disabled cattle from the human food chain; prohibiting air-injection stunning of slaughter cattle; requiring additional process controls in advanced meat-recovery systems; and forbidding the use of mechanically separated meat in human food. [2] Since 1990, the APHIS has conducted disease surveillance targeting cattle populations where the disease is most likely to be found. Surveillance in the United States has increased steadily from 1990 and jumped significantly in 2004 when USDA implemented enhanced surveillance following the detection of BSE in 46 an imported cow in December 2003. The USDA has sampled more than 759,000 animals and, to date, only 2 animals have tested positive for BSE under the program. [8] The surveillance has included the predeath inspection of slaughter cattle and prohibiting of animals with any clinical signs of neurological disease or other abnormalities. [2] Besides extensive surveillance, preventative measures also include education. The APHIS educates veterinary practitioners, veterinary laboratory diagnosticians, industry, and producers about the clinical signs and pathology of BSE. Videotapes of cattle showing clinical signs of BSE and BSE factsheets, risk assessments, and reviews have been widely distributed to State and Federal veterinarians, private practitioners, other industries, and producers. APHIS is continuing an education effort to inform U.S. cattle producers and veterinarians about this disease. Numerous briefings have been held for industry groups. In addition to press releases and factsheets, a videotape on BSE and an information packet were distributed to all APHIS field offices, state veterinarians, extension veterinarians, colleges of veterinary medicine, and industry groups. [2] The World Health Organization (WHO) has released an extensive list advising nations and industries about prevention. This included prohibiting the use of ruminant tissues in ruminant feed and the exclusion of tissues that are likely to contain the BSE agent from any animal or human food chain. [6] Also, the WHO states that “all countries are encouraged to conduct risk assessments to determine if they are at risk for BSE in sheep and goats.” *6+ Reassurance of meat being safe can be provided by removal of visible nervous and lymphatic tissue from meat (skeletal muscle). [6] The WHO has also advised the pharmaceutical industry to avoid the use of bovine materials and materials from other animal species in which TSEs naturally occur and that if absolutely necessary, bovine materials should be obtained from countries which have a surveillance system for BSE in place. [6] REFERENCES 1. Centers for Disease Control [homepage on the internet]. Atlanta, GA: The CDC; 2009. [cited 2010 Mar 16]. BSE (Bovine Spongiform Encephalopathy, or Mad Cow Disease). Available from: http://www.cdc.gov/ncidod/dvrd/bse/ 2. Bovine Spongiform Encephalopathy: An overview. USDA. 2002 Dec. [Online] [Cited March 17, 2010]. Available from: http://www.aphis.usda.gov/publications/animal_health/content/printable_version/BSEbrochure122006.pdf 3. Kimberlin, R. Bovine Spongiform Encephalopathy [book on internet]. Edinburgh, UK: Food and Agriculture Organization; 1993 [cited 2010 Mar 16]. Available from: http://www.fao.org/docrep/T0573E/t0573e00.htm 47 4. BSEinfo [homepage on the internet]. Beef Checkoff; 2009 [cited 2010 Mar 16]. BSE. Available from: http://www.bseinfo.org/scieBSE.aspx 5. World Organization for Animal Health [homepage on the internet]. Paris, France: OiE; 2002 [cited 2010 Mar 16]. Bovine Spongiform Encephalopathy. Available from: http://www.oie.int/eng/maladies/fiches/a_b115.htm 6. World Health Organization [homepage on the internet]. Switzerland: WHO; updated 2002 [cited 2010 Mar 16]. Bovine Spongiform Encephalopathy. Available from: http://www.who.int/mediacentre/factsheets/fs113/en/ 7. United States Department of Agriculture [homepage on the internet]. Atlanta, GA: USDA; 2005 [cited 2010 Mar 16]. Food Safety and Inspection Service: Facts Sheet: Bovine Spongiform Encephalopathy. Available from: http://www.fsis.usda.gov/FactSheets/Bovine_Spongiform_Encephalopathy_Mad_Cow_Disease/index.as p 8. Animal and Plant Health Inspection Service [homepage on the internet]. Washington, D.C.: APHIS; 2006 *cited 2010 Mar 16+. USDA’s BSE Surveillance Efforts. Available from: http://www.aphis.usda.gov/publications/animal_health/content/printable_version/fs_BSE_ongoing_vs. pdf 48 HERPES: A Sexually Transmitted Disease Courtney Tipper General Microbiology, Spring 2010 INTRODUCTION Sexually transmitted diseases (STDs), once called venereal diseases, are among the most common infectious diseases in the United States. This is because many people in the United States of America prefer to ignore the statistics about STDS. Understanding the basic facts about STDs, including the ways in which they are spread, their common symptoms, and how they can be treated is therefore the first step toward prevention. The incidence of STDs is on the rise, in part because in the last few decades, young people are becoming sexually active earlier, and yet are marrying later. In addition, divorce is more common. The net result is that sexually active people today are more likely to have multiple sex partners. Another factor is that some STDs tend to have no symptoms, and are therefore easily spread by asymptomatic individuals. One such disease is HERPES, a common disease especially on college campuses. DESCRIPTION Herpes is a sexually transmitted disease caused by the Herpes simplex viruses type 1 (HSV-1) or type 2 (HSV-2). HSV 1 is commonly known as oral herpes, and HSV 2 is commonly known as genital herpes. Herpes simplex type 1 causes sores around the mouth and lips (sometimes called fever blisters or cold sores). HSV-1 can cause genital herpes, but most cases of genital herpes are caused by Herpes simplex type 2. In HSV-2, the infected person may have sores around the genitals or rectum. Although HSV-2 sores may occur in other locations, these sores usually are found below the waist. Viruses that enter the body often go through a latency period. A latency period is a stage during which the virus is dormant and inactive. Therefore, there are no external symptoms that the virus is in the body. At some point, however, the virus becomes active again. Any number of factors can cause reactivation of the virus including, physical or emotional shock. When the virus is reactivated, symptoms of the infection reappear. This pattern explains why cold sores and genital herpes commonly appear and then disappear. Each new appearance does not mean a new infection. It means that the virus has emerged from its latency period and become active. SIGNIFICANCE In the United States, more people have genital herpes than all other sexually transmitted infections combined, 50 million people in total. [1] Additionally, there are about one million new genital herpes infections each year attributable to HSV-2. 80-90% of people who have genital herpes report no history of signs or symptoms consistent with genital herpes. The National Health and Nutrition Examination Survey show that only 14.3% of those testing positive for HSV-2 are aware that they have genital herpes. Therefore, asymptomatic individuals can continue to engage in unprotected sexual activities that facilitate the spread of HSV to non-infected individuals. Both HSV-1 and HSV-2 cause life-long infections; however, the natural history of genital infection is substantially different for the two types. About 10% of the population acquires Herpes infection through the genital route and the risk is concentrated in 49 young adulthood. Primary infections of all kinds are rare after age 30, thus making this disease significant for college students who may be leaving home for the first time and initiating their first relationships with individuals of the opposite sex. Following infection, 45% of orally infected individuals and 60% of patients with genital herpes will experience recurrences. [1, 2] The lesions of oral herpes are prominent on the lips and therefore create embarrassments for those afflicted by the virus. Ocular herpes is also responsible for 3% of all primary herpes infections. The recurrence rate for ocular herpes is 40%, and in the United Kingdom alone, there are 50,000 cases per year. [3] The incidence of neonatal herpes is about 1 in 4,000 live births with 70 deaths per year in the United States. [4] This shows that herpes is a disease that affects everyone at every age, and once infected, reoccurrence is common. PATHOGENESIS The pathogenesis of Herpes depends on the ability of HSV to escape the immune response, and persist indefinitely in a latent state. The exact mechanism of latency is not known; however, the virus may remain in a non-replicative mode, and be maintained within the cell by integration into the cellular chromosome. [4] The virus may also persist in certain tissues by a mechanism of controlled low grade productive virus infection that does not lead to cell lysis. Both HSV-1 and HSV-2 can also infect macrophages and lymphocytes. In these cells, the virus creates syncytia, resulting in the formation of giant cells and the rapid destruction of the affected cells. The virus can also escape the immune system by coating itself with IgG via Fc receptors and complement receptors. HSV can also escape the immune system by spreading from cell to cell without entering the extracellular space and coming in contact with humoral antibodies. [5] GENERAL SYMPTOMS Once individuals have been exposed to the virus through direct skin-to-skin contact, it can take anywhere from two to 20 days before the first episode of symptoms of genital herpes will occur. While the first outbreak of herpes is usually the worst that one will experience, as much as 60% of those infected with the virus will fail to notice any symptoms because the symptoms are so mild. Even if one does not have any symptoms, they can still pass on the virus to their sexual partners. On average, the first herpes outbreak in men lasts about two weeks while in women the average is three weeks. Additionally, because women frequently experience their herpes sores in the vagina, many may mistake the few herpes symptoms they have for some other type of infection. People may also confuse herpes with an outbreak of boils. Boils are larger and more painful; but, it is not uncommon for a woman infected with the herpes virus, to be misdiagnosed with yeast infection, pelvic inflammatory disease, or an inflamed cervix. While women frequently develop vaginal herpes, it is also possible to develop lesions on the vulva, cervix, urethra, anus, thighs and buttocks. If a woman has unusual vaginal discharge or persistent vaginal discomfort, it is a good idea to be tested for herpes. The most common sign of herpes is red sensitive skin that develops sores or blisters. These lesions usually show up in and around the genital area, although female symptoms of herpes may cause these blisters to develop inside the vagina. Other signs and symptoms of genital herpes include swollen lymph glands in the groin, headaches, muscle aches, fever, and lower back pain. In women, herpes symptoms can also include vaginal discharge and pain, or a burning sensation when urinating. About 25% of women will develop meningitis as a complication of their first herpes outbreak while another 10% to 15% will have trouble urinating 50 because of their herpes. Of those people who experienced an outbreak of herpes sores when they were first infected, 80% will go on to have recurrent outbreaks. Although these outbreaks tend to be less severe and shorter in duration than the first outbreak, they can still be just as uncomfortable. Additionally, a person is more likely to pass on the virus when they experience an outbreak. Prior to the recurrent breakout, many individuals tend to experience warning symptoms that they are about to have another herpes episode. Often, an individual will have some pain or discomfort in the area that was infected before their herpes sores developed. During this time, infected persons are considered to be very contagious despite the absence of herpes blisters. DIAGNOSIS Laboratory tests can be used to detect the virus directly from skin lesions. A swab is rubbed over the sore and the material on the swab is used for one of several tests including culture, FA, or PCR. Serology can show evidence of antibodies in the bloodstream to herpes infection. Both virus detection and serology can help confirm the suspicions from physical examination and history that infection with genital herpes may have occurred. Although genital herpes is common, it is difficult to diagnose just by physical examination and history. An accurate laboratory test should therefore be utilized to establish the diagnosis. Testing for the virus directly from the skin is useful if genital symptoms are present. Diagnosing herpes by just looking at a lesion or sore does not give an accurate diagnosis because many other infections or irritations can look just like herpes. As such, and because HSV is a chronic disease, confirmation of the diagnosis with a laboratory test is recommended by almost all medical authorities and by the Centers for Disease Control. When lesions or sores are present, the physician can swab the sore and submit the sample for detection of Herpes simplex virus by growth in culture, or by detecting parts of the virus in the material on the swab. Finding the HSV virus by any of these assays shows that HSV caused the lesion. Another way to diagnose herpes is to detect antibodies in the blood that the body has made to fight off the virus after infection. These antibodies last for a lifetime after an infection. They can be detected by a blood test even if no signs or symptoms are present at the time the patient visits the doctor. Antibodies develop even when patients have never had symptoms of herpes. Serology can help show that herpes infection has occurred in situations where it is difficult to obtain or to properly transport a sample for virus detection tests. If performed correctly, antibody tests can identify a person as having had an HSV-2 or an HSV-1 infection, or both infections sometime in the past. TREATMENT Unlike other sexually transmitted diseases, herpes cannot be cured because medication that will attack the virus while it lies dormant in the nerve cells will also damage the nerve cells. However, there is treatment available for acute infections or an outbreak that involves the use of anti-viral drugs such as Acyclovir, Valaclovir or Famcyclovir. Acyclovir has been found to reduce the reproduction of the virus in initial outbreaks, thus possibly lessening the number of subsequent outbreaks. To be effective, therapy must be started immediately after the first sores appear. Every sexual partner of the infected person needs to be examined, and if necessary, treated. Famcyclovir and Valaclovir have similar effects and may work to prevent a herpes infection from establishing itself if taken soon enough in the course of the illness. Long-term drug therapy (suppressive treatment) may be helpful for individuals who suffer frequent recurrent outbreaks. Suppressive treatment will reduce outbreaks by 85 percent and reduces 51 viral shedding by more than 90 percent. Topical antibiotic ointments also may be applied to prevent secondary bacterial infections. PREVENTION During an outbreak of genital herpes, a number of measures can be taken to make the patient more comfortable such as: wearing loose clothing, avoiding excessive heat or sunlight, keeping the sore area clean and dry, and placing cool or lukewarm cloths on the sore area for short periods of time. Patients should not use perfumed soaps, sprays, feminine deodorants, or douches. They should also take aspirin, acetaminophen or ibuprofen for the pain, avoid touching sores but wash hands if the sores are touched. Because the chances of contracting this disease increase with the number of sexual partners a person has, limiting the number of partners is the first step toward prevention. Many couples have had sexual relations for years without transmitting herpes. Some simply avoid having sexual contact when signs or symptoms are present. The risk of transmitting the virus may possibly be reduced if one uses condoms. The continued use of condoms in a long-term relationship is a personal decision that only the couple can make. However, at all costs, couples should try to avoid sexual intercourse during an active episode of herpes, because this is when the virus is most likely to be transmitted. This period includes the time from when a partner first has warning signs of an outbreak, such as a tingling or burning in the genitals, until the last of the sores has healed. Also, sexual activity prolongs the healing of the episode. Transmission risk is increased if there are any breaks in the skin, for example, if one has thrush or small abrasions from sexual intercourse, often due to insufficient lubrication. It can be helpful to use a lubricant specifically for sexual intercourse and to avoid sex if one has thrush. Sexual lubrication is helpful right at the start of sexual activity. Sores in other areas, such as the buttocks and thighs, can be just as contagious as those in the genital area, and care should be taken to avoid direct contact with such sores during sex. At other times, there is still a small risk of transmitting the infection, even if there are no signs of genital herpes. REFERENCES 1. U.S. Department of Health and Human Services (2006). Genital HSV infections. Sexually Transmitted Diseases Treatment Guidelines 2006 (CDC Publication Vol. 55, No. RR-11), pp. 16-20. Atlanta: U.S. Department of Health and Human Services. [Online] [Cited March 16, 2010] Available from: http://www.cdc.gov/std/treatment/2006/rr5511.pdf 2. Corey L Herpes simplex virus. In GL Mandell et al., eds., Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. 6th edition, vol. 2. Philadelphia: Elsevier; 2005. pp. 1762- 1780. 3. Hill TJ, Field HJ, Blyth WA. Acute and recurrent infection with Herpes simplex in a mouse: a model for studying latency and recurrent disease. Journal of General Virology. 1975; 61: 341-353 4. Pathogenesis of Herpes simplex virus infection. [Online] [Cited March 17, 2010]. Available from: http://virology-online.com/viruses/HSV2.htm 5. Hunt R. Herpes virus. [Online] http://pathmicro.med.sc.edu/virol/herpes.htm [Cited 52 March 17, 2010] Available from: Borrelia burgdorferi Jolie Wax General Microbiology, 2010 DESCRIPTION Borrelia burgdorferi is a bacterium that belongs to the phylum of Spirochaetes. It is long, thin and helically coiled, with axial filaments that run lengthwise between the peptidoglycan layer and the outer membrane. It is commonly mistaken as being Gram-negative due to the double membrane envelopes; however, the envelopes have significant differences in their composition and architecture. [1] The bacteria move in a screw-like motion produced by the flagellum. The flagella or axial filaments also determine the shape of the organism. Borrelia cells are an average of 0.2 to 0.5 m. [2] The single linear chromosome is about 900 kbp and the cells contain 12 linear and 9 circular plasmids. The strain that has been fully sequenced and used widely in laboratories is named B31. Growth of B. burgdorferi involves some unique nutritional requirements. Spirochetes in general are considered chemo-heterotrophic and B. burgdorferi can be anaerobic or microaerophilic. They require carbohydrates as a source of carbon and energy. [3] Culturing these bacteria in vitro has posed some difficulty partially due to differentiation associated with gene expression. They are also fastidious and require low oxygen and specially enriched media. Lab cultures over time change protein expression, lose plasmids and their ability to infect an animal host. This suggests the importance of the microclimate, temperature and pH, provided by the required Ixodes ticks that propagate the bacteria in the gut. [4] This mechanism also suggests the evolution of B. burgdorferi with the arthropod host. [5] Despite the large number of plasmids, the overall genome size of B. burgdorferi is small and has an absence of genes for the synthesis of amino acids, fatty acids, enzyme cofactors and nucleotides. For these reasons, B. burgdorferi requires serum in supplemented mammalian tissue culture medium. Glucose, fructose and disaccharides are acquired by the phosphotransferase system and used in glycolysis. Nacetylglucosamine must be added to culture medium for the growth of B. burgdorferi. [4] A complex liquid medium called Barbour-Stoenner-Kelly can be used to culture the bacterium. [7] The genomic expressions of this organism alter significantly depending on its environment. SIGNIFICANCE B. burgdorferi is one of three known species of Borrelia spirochetes that cause Lyme disease. Lyme disease is characterized by skin lesions, flu-like symptoms, neurological abnormalities and arthritis. It is the most common tick-borne illness, and in the United States, it is recorded at 17,000 cases per year. [3] Lyme disease affects adults and children alike. Other sources such as the Centers for Disease Control (CDC), estimate 200,000 new cases per year, with a total of 1.8 million Americans infected. [6] The disease primarily occurs in northeastern and mid-western states although it has appeared in western parts of California and Oregon. It has been reported in many temperate parts of the northern hemisphere and continues to spread. [1] The disease is prevalent in Europe and Asia as well. In the 53 United States the cost is estimated to be $2.5 billion over 5 years. This is due to the fact that it is difficult to diagnose. In some regions including New York and Connecticut, Lyme disease has caused significant depreciation of real estate values. [7] The disease is easy to treat if the diagnosis is made on time; however, due to the difficulty in recognizing its symptoms, and therefore, starting treatment quickly, the disease can progress to a chronic, debilitating stage with grave and complicated outcomes. PATHOGENESIS The mechanisms of Borrelia transmission and infection are very complex and involve several host and ecological conditions. The most prominent mechanism is known as an enzootic life cycle involving the arthropod vector and the mammalian host. The tick, Ixodes ricinus, is the most common vector, but Borrelia may also be transmitted by fleas, mosquitoes and mites. A variety of mammalian hosts include field mice, white tailed deer, dogs, cows and humans. Some mice and other animals make poorer hosts because they do not remain carriers for extended periods. [8] They are unique among pathogenic spirochetes because they require an obligate blood feeding arthropod for transmission and maintenance in vertebrate hosts. [1] If the tick has not fed on an infected mammalian host, it is not able to transmit the disease. The bacteria remain in small numbers in the tick gut until blood feeding begins, and they rapidly multiply in the gut. After two days of feeding, the bacteria move into the saliva of the tick and infect the mammalian host. It has been speculated and with evidence that the bacteria may spread through sexual intercourse and from mother to child in-utero. Research at the University of Wisconsin found that dairy cattle can acquire the disease and pass it to humans via the food chain. [6] An important aspect in the course of transmission is the differential gene expression of the outer surface proteins (Osps) of Borrelia. The presence of major surface lipoproteins at the host-pathogen interface, ensures pathogenicity. Within the tick, a protein called ospA is highly expressed. This protein is responsible for enabling the bacteria to anchor to the midgut cells and avoid phagocytosis. [2] Upon migration to the saliva during feeding, the OspA is down-regulated and often absent while an OspC becomes highly expressed during transmission. OspA is not found within the mammalian host. OspC can fall below detectable levels as the infection persists and reappears later. Thus, a two-step adaptation to a mammalian host has been repeatedly observed. In adjustment to the tissue microenvironment, less than 40 of the 137 lipoprotein genes are expressed, followed by 116 genes being actively transcribed within ten days of infection. [2, 5, 7, 8] Dissemination from the area of the tick bite is due to the causative agent attaching to host plasmin, and causing degradation of glycoproteins. [7] A group of 80 lipoprotein genes, including OspC, are persistently expressed during infection when no immune pressure is present and down-regulated under immune pressure. It takes 5 weeks for B. burgdorferi to become host adapted to immune selection pressure. A third group of lipoproteins are expressed throughout mammalian infection. [5] Through gene regulation and unique outer surface proteins, B. burgdorferi is able to establish chronic infections. There are several linear and circular plasmids known to have a role in persistent mammalian infection. The Ip25 plasmid contains a gene that encodes a nicotinamidase that functions in the synthesis of NAD. This appears crucial for growth within a host. A similar one is named Ip28-1. When both plasmids are 54 lost in the case of in-vitro growth, there is no change in growth ability, but they are unable to cause infection in immune deficient mammals. A series of circular plasmids named cp32’s (prophage genomes), play a role in horizontal DNA transfer among B. burgdorferi. [2] Furthermore, the spirochete’s motility and chemotaxis mechanisms play a role in its ability to colonize multiple tissue sites and enter the bloodstream. SYMPTOMS The role of Osps is not only in transmission between hosts, but also in the bacteria’s ability to infect the host and produce an inflammatory immune response. Symptoms of B. burgdorferi range from a localized rash, erythema migrans, to nueron demyelination, damage in the myelin sheath of the neuron. [3] Some sources note that the disease is caused by the hosts’ immune response and not mediated by toxigenic molecules. [1] Other research has identified specific biotoxins from B. burgdorferi. [6] Lyme disease presents itself in three major stages. Within ten days of an infected tick bite, a red ring with a central clearing, erythema migrans, may develop at or near the site of the bite. This may be asymptomatic, or itch and burn, and is 80% of the time accompanied by flu like symptoms, including headaches, fever and fatigue. If this remains untreated, it can last for 2-3 weeks. 20% of patients experience this recurrently. The second stage, once the bacteria have disseminated, occurs weeks to months after infection. Patients may experience neurological abnormalities, heart inflammation and arthritis usually in the elbows and knees. Lyme arthritis is not always accompanied by inflammation, and recurrent episodes may last up to ten years. 10% of these patients will have chronic arthritis. The last stage may appear years later, in which infected individuals experience neuron demyelination accompanied by symptoms similar to Alzheimer’s disease and multiple sclerosis. [3,8] Other symptoms include chronic neuropathy, cranial nerve palsies, meningitis and chronic fatigue syndrome. [8] DIAGNOSIS Laboratory diagnosis of Lyme disease is very difficult because B. burgdorferi adapts and migrates quickly and can avoid the immune cells. Lyme disease is too often misdiagnosed in part because erythema migrans only develop 30% of the time. [6] When this is the case, a biopsy should be performed immediately to confirm the presence of B. burgdorferi. Blood tests can only be accurate 4-6 weeks after infection and are still notoriously inaccurate. It is very common for patients to have had a tick bite with continual symptoms and still produce negative laboratory results. The most common blood test done to detect Lyme disease is called the ELISA test and costs about $60, and is very inaccurate. It tests for the antibodies produced in response to B. burgdorferi infection. [9] Serum antibody and immunoglobulin IgM and IgG tests can also be unreliable because of the bacteria’s ability to hide outside the bloodstream. The association between B. burgdorferi and chronic toxin mediated illness has lead to some advancement in detecting it. This is because available data suggest that even after apparently killing the bacteria with antibiotics, symptoms still persist. Biologically produced organic neurotoxins can be effectively identified by a contrast sensitivity test (CS). CS is used repeatedly with cholestyramine treatment to measure improvement and therefore provide the necessary “bio marker” for neurotoxins. [9] Patients with chronic Lyme can take a CS test called FACT assay which only takes 5 minutes, is inexpensive and can even be done online. This test involves the brain’s ability to distinguish between 55 black, white and grey. This method is effective for chronic Lyme patients because the eyes show the greatest susceptibility of the whole body to acute and chronic effects of neurotoxins. The treatments prescribed for Lyme disease reflect the stage of the disease, and an accurate diagnosis. Prevention however, is the best medicine and the only fool-proof method to not living with the disease. PREVENTION There is a protocol for the natural prevention of Lyme disease as well as the development of vaccines. Strategies for the prevention of Lyme disease are the prompt removal of ticks using tweezers to extract the head. This is important because it takes 24 hours for the bacteria to be transmitted. [3] Humans are infected by the ticks in the nymph stage, from May to July, and this is when 85% of people become infected. The other 15% are infected in the fall when ticks are adults. [8] It is recommended that if you go into the woods at this time, to wear light colored clothes and tuck your pants into your socks, wear a hat and check all clothing and skin when you come out of the woods and even the tall grass. Over the counter repellents are available. Premethrin kills ticks on contact and is approved for use on clothing only. Repellents containing high concentrations of diethyltoluamide are also effective. [3] Various vaccinations have evolved since the discovery of Lyme disease. The first one approved for human use in 1998, is LYMErix. [7] The vaccine is composed of lipidated recombinant OspA and three doses are recommended at 1 month intervals with booster shots every spring. This vaccine however protects against Borrelia burgdorferi , but not other subspecies. LYMErix is approved for people of the ages 15 to 70. The vaccine’s usefulness is very limited because of the age requirements and the cost-effectiveness. It is also linked with severe side effects, consequently, the vaccine is no longer recommended for use in the prevention of Lyme disease. TREATMENT Antibiotic treatment of Lyme disease is most effective when it is used in the first stage. Patients given amoxicillin, 500 mg three times a day, or tetracycline have a rapid recovery with no arthritis. Doxycycline is also often prescribed in the early stage of infection, 100 mg twice a day for 28 days. Several months of antibiotics are not uncommon. Treatment with antibiotics is considered 90% effective when used within 4-6 weeks. [7] If the nervous system becomes involved, cerfiaxone is used because it can cross the blood brain barrier. [3] In cases of chronic Lyme disease, alternative medicine approaches are arguably more successful. Patients are often placed on nutritional diet of anti-inflammatory foods such as fish oil and borage seed oil. Calcium and magnesium can help with muscle pain. For patients placed on long term antibiotic therapy, it is important to also take probiotics to maintain a balance of intestinal flora. [6] Finally the treatment used in conjunction with the contrast sensitivity test is cholestyramine. This drug is used for many other toxin mediated illnesses because of its ability to flush circulating toxins out of tissues, molecule by molecule. [9] Cholestyramine leaches cholesterol out of bile in the small intestine and can do the same thing with other substances that move through the gall bladder and fatty tissues. 56 Contrast sensitivity and FACT scores show a dramatic improvement and rapid reduction of painful symptoms. [9] There is no definite cure for Lyme disease once the bacteria have disseminated after approximately ten days and organic toxins are being produced. If someone plans on spending any time near or in the woods, it is incredibly important that they understand how to protect themselves. This is a growing disease that often goes underestimated, overlooked and misdiagnosed. DISCUSSION Lyme disease interests me for many reasons, one being that I spend a lot of time outdoors, and I know too many people afflicted with this disease. In microbiology, I find great interest in organisms that are on the forefront of evolving toxins, possibly from our growing and expanding chemical world. Pollution and encroachment is not ruled out as a cause of this disease and this correlation is being researched today. The Borrelia spirochete is highly adaptive to multiple environments and hosts. It would be incredibly interesting to understand more about its co-evolution with specific species or arthropods and mammals, and the immunity of some animals that carry or expel the bacteria with no known symptoms. Many symptoms of chronic Lyme are consistent with health issues that afflict so many people such as; chronic fatigue, sick building syndrome arthritis, depression and chronic immune depressed diseases. Continuing research in the differential gene expressions, due to environment, host, available nutrients and pollution with B. burgdorferi will be useful because the B31 strain has been fully sequenced including the 20 plasmids. This research is also necessary because of Borrelia’s recent migration into our lives. More recent research shows that the bacteria can survive and be transmitted in an environment without the arthropod host, and within a mammalian population. Research using Borrelia may give insight to the development of medicines for other chronic ailments with symptoms that are congruent with Lyme disease. Microorganisms are ubiquitous in the environment and our bodies, and have the ability to adapt and mutate with high frequency. Our society and the human race in general seem to be plagued more and more with chronic and neurological disorders. We are also dramatically changing and polluting our environment. The connections between these two statements can be somewhat explained by the role of microorganisms, and research in this area will contribute to necessary advancements for protecting human health. REFERENCES 1. Samuels DS, Radolf JD. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk, England: Caister Academic Press; 2010. 2. Brown University. Lyme Disease. [Online] [accessed 17 March 2010]. Available from: HYPERLINK http://www.brown.edu/Course/Bio_160/Projects2005/lyme_disease/organism.htm 3. Willy J, Sherwood L, Woolverton C. The Deinococci and Gram-Negative Nonproteobacteria. Prescott’s Principals of Microbiology. NY, New York: McGraw Hill; 2000. p. #.[435-436] 4. Fraser CM, Casjens S, Huang WM., et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997; 390: 580-586 [accessed 17 March 2010]. Available at: 57 http://HYPERLINK "http://www.nature" www.nature.com/nature/journal. 5. Liang FT, Nelson FK, Fikrig E. Molecular adaptation of Borrelia burgdorferi in the murine Host. Journal of Internal Medicine. 2002; 196 (2): 275-280 6. Lyme Disease. [homepage on the web] [accessed 18 March 2010]. Available at: HYPERLINK "http://eiresources.org/illness" http://ei-resources.org/illness information/related-conditions/lyme-disease. 7. Thanassi WT, Schoen RT. The Lyme disease vaccine: conception, development, and implementation. Annals of Internal Medicine. 2000; 132 (8): 661 - 668 8. Meyerhoff JO. Lyme Disease: eMedicine Rheumatology. 2009. Department of Internal Medicine, John Hopkins University School of Medicine. [Online] [accessed 17 March 2010]. Available from: HYPERLINK "http://emedicine.medscape.com/article/330178-overveiw" 9. Shoemaker Ritchie C. Desperation Medicine. Baltimore Maryland: Gateway Press Inc.; 2001. 58 Malaria Julia Greenfield General Microbiology, Spring 2010 GENERAL DESCRIPTION OF DISEASE Malaria is a febrile illness that is commonly transmitted by infected mosquitoes. Most infections occur in tropical places, and symptoms can range from mild flu-like symptoms to severe malaria, and even death. The disease is not caused by the mosquito, but rather a parasite uses the mosquito as a vector. The parasite is transferred through the saliva of an infected mosquito to a host. The parasite grows and reproduces in the host, causing destruction of red blood cells and the symptoms associated with the disease. [1] SIGNIFICANCE As early as the fifth century BCE malaria had been recognized through its characteristic episodes of chills and sweats. The name comes from the Italian phrase mal’ aria meaning “bad air” because it was linked to swamps; however, 17th century Italians did not make the connection to the mosquitoes that lived there. [2] The disease has plagued mankind ever since and today, malaria is commonly transmitted throughout most of the tropical regions of the world, and almost half of the world’s population is at risk of being infected. [3] Malaria is especially a problem in parts of sub-Saharan Africa, southern Asia, and northern South America. The World Health Organization believes that over 200 million people become sick enough from malaria to seek medical help each year. [4] While this number is staggering, it pales when it is realized that in Africa, 80% of malaria cases are treated at home. [3] In 2008, between 700,000 and 1 million deaths were caused by malaria. [4] Almost 90% of all malaria related deaths occur in Africa, making it the second leading cause of death by infectious disease behind HIV/AIDS. [1] In countries where malaria is prevalent, the disease demands a significant portion of the public heath expenditures, as high as 40%. Infected individuals also lose income from sick days, cost of medication and other heath-related costs. Malaria prevention and treatment account for as much as 50% of hospital admissions and 60% of outpatient health clinic visits in these same countries. [4] It is estimated that over 12 billion dollars of Africa’s valuable economic dollars are spent on malaria each year. [3] Malaria has wreaked havoc on mankind for many years, taking a tremendous human and economic toll. In spite of substantial research funds, no preventive vaccines are in sight; however, the parasites are acquiring resistance to anti-malarial drugs. SYMPTOMS Malarial symptoms can be divided into three categories. The first category is the cold stage where the infected person experiences shivers and chills. In the hot stage the host may display fever, headaches, vomiting and even seizures. The last category is the sweating stage where the host sweats, feels tired, and has a reduction in fever. Other symptoms include; nausea and body aches. Signs of malaria include increased respiratory rate, some jaundice, metabolic acidosis, hypoglycemia and enlargement of liver and or spleen. [1] 59 Malarial paroxysms are caused by the periodic release of erythrocyte cell fragments and merozoites into the bloodstream after the lysis of infected erythrocytes. After several waves of paroxysms, the parasite may go into remission for several weeks to months. Other symptoms include anemia, where the blood does not have enough oxygen for the body. [5] The anemia associated with malaria is due to the destruction of erythrocytes. According to the CDC, lysed red blood cells release “hemozoin and other toxic factors such as glucose phosphate isomerase (GPI)” which stimulate other cells to produce cytokines and induce the high fevers associated with the disease. [1] Lysed erythrocyte cells cause hemoglobinuria (hemoglobin in the urine) and even kidney failure. In addition, erythrocytes infected with malaria in the trophozoite stage have a tendency to stick to the endothelium of blood vessels and inhibit the flow of blood. In cerebral malaria, the clumping of erythrocytes occurs in blood vessels in the brain, which can lead to abnormal behavior, seizures, coma, and other neurological symptoms. [1] Depending on the type of Plasmodium present and the health of the host, malaria can present with a combination of the symptoms above. DISEASE CAUSING AGENT Protists are a diverse group of eukaryotic microorganisms that may have characteristics of both plant and animals, but is not considered either; they can be unicellular or multicellular. [6] The structure of a protist varies depending on the type of organism. Plasmodium is a genus in the kingdom of protist and it is a single celled organism. It is of the subclass, coccidia and produces sporozoans. [7] All species of Plasmodium will go through a variety of structural changes depending on where it is in its life cycle. It can go from having one nucleus to several when it is in its growing stage. Plasmodia have 14 chromosomes and one mitochondrion, but few other membrane-bound organelles. Malaria is caused by organisms in the Plasmodium genus, more specifically, P. falciparum, P. malariae, P. ovale, and P. vivax. [2] P. falciparum is considered the most dangerous of the malarial causing parasites because it is linked to the most severe cases of the disease. It is estimated that 90% of all deaths attributed to malaria are caused by this parasite, as well as 91% of all infections. The species is found most commonly in Africa but exists in smaller populations elsewhere. Its high virulence is due to the fact that it produces the most merozoites in a day, up to 40,000, and is prone to clumping in the trophozome stage. Its resistance to chloroquine thwarted efforts to eradicate malaria in the 1960’s. Regarded as the least dangerous, is P. malariae though it is more common than P. falciparum. It has lower merozoite counts and a low rate of morbidity. It has an unusually long incubation period of 16-56 days. Unlike the other three species of parasites, P. malariae hosts have fevers that occur every three days, while the others have fevers every two days. P. ovale and P. vivax are very closely related, and specific to P. ovale is its ability to persist in the liver for long periods. P. vivax is found mainly in Asia and Latin America. It has a high tendency to cause an enlarged spleen but is not often fatal. [8] PATHOGENESIS The life cycle of the malarial causing parasite begins when the parasite is introduced into the host’s bloodstream. In this case it will be transferred from an infected female Anopheles mosquito to a human host via a mosquito bite. During the bite, the mosquito uses its maxillae to pierce the human skin and as it feeds, it injects the infected saliva into the host. [9] The salvia contains an anticoagulant to aide in 60 blood flow to the insect and also small Plasmodium sporozoites. These sporozoites travel through the bloodstream until they reach the liver and enter the hepatic cells. They remain in the liver and undergo schizogony; multiple asexual fission. [2] Here, the parasite may remain dormant in the incubation phase, which lasts from one week to about a month, before any symptoms manifest. [1] After a critical point is reached, the newly formed merozoites are released into the blood. The merozoites in the blood come into contact with erythrocyte cells and enter them. Inside the erythrocytes, the parasite increases in size and becomes a trophozoite, which is a cell with one nucleus. As the nucleus divides to produce 6 to 24 nuclei, it is called a schizont. This phase continues into the next phase where the schizont divides to produce many mono-nucleated merozoites. At the critical point, the erythrocyte will lyse and release many merozoites into the bloodstream to infect more erythrocytes. This process will repeat itself in intervals; the time range between each cycle can be as short as every two to three days or longer depending on the species of malaria causing Plasmodium. [1] Intermittently, the Plasmodium infection can be temperate and will not lyse the infected erythrocyte of the human host; instead it will produce microgametocytes and macrogametocytes which are the male and female gametocytes respectively. The microgametocytes and macrogametocytes can travel through the bloodstream and can be consumed by a mosquito during a bite. The mosquito is then infected and the infected microgametocytes and macrogametocytes further differentiate to produce male and female gametes which eventually lyse the erythrocyte and undergo fertilization in the stomach of the mosquito. The fertilized zygote is called an ookinete which attaches and gains entry into a cell of the mosquito stomach. In the stomach cell, the ookinete develops into an oocyst and penetrates the stomach wall. Outside the stomach, the oocyst divides by meiosis to form sporozoites which migrate to the salivary glands of the mosquito. Here they are primed to begin the life cycle again once the mosquito feeds on another vertebrate host. [1] Transmission of malaria from mosquito to vertebrate host is determined by several factors. One factor is climate, usually more tropical climate with regular rainfall is ideal for the population of Anopheles mosquitoes. A warmer temperature above 77oF increases the activity of the mosquito and thus increases the chance of an infection. Behavioral differences among some species of female Anopheles mosquitoes will determine which host the mosquito will feed from in order to gain a blood meal to facilitate egg formation. Of the 3,500 species of mosquitoes, only 30-40 transmit malaria. Anopheles mosquitoes that prefer to feed on humans are called anthropophilic, and will have more chance to transfer the parasite to a human. Malaria can also be transmitted through infected blood, in a blood transfusion and to newborns during birth if the mother has the disease, though this is rare. [1] Individuals that are heterozygous for the sickle cell allele are partially protected from malarial infection by the parasite. There are two explanations as to why individuals with sickle cell allele are protected. The sickle cell allele denoted as Hb-S causes the red blood cells to be abnormally shaped into a crescent shape. The abnormal hemoglobin of a person with the sickle cell trait will cause the red blood cell to become sickle shaped when infected with the malaria parasite. The spleen then separates these abnormally shaped blood cells out of the bloodstream and destroys them, and the parasite is killed 61 along with the red blood cell. [10] Another theory is that the sickle shape of some erythrocytes will cause low oxygen binding, decreasing the oxygen level in the blood. The Plasmodium parasite is an obligate aerobe that will not survive in the decreased oxygen levels of the abnormal blood. [2] In some areas where malaria is common like certain parts of Africa, as much as 40% of the population has one Hb-S gene. [10] A study in Kenya found that the sickle cell trait provides 60% protection against overall mortality within the first 16 months of life. [1] DIAGNOSIS Malaria is often misdiagnosed as the flu or a cold in countries where malaria is uncommon. In parts of the world where malaria is common, it is often self-diagnosed by recognizing the symptoms. Laboratory diagnosis of malaria is made by the observation of the parasite in blood samples via a microscope. Using thick smears and Romanowsky stains, the presence of malaria can be diagnosed more effectively when examining red blood cells. A thin smear is good to identify malaria species present. The Quantitative Buffy Coat (QBC) Test is a new and fast way to diagnose a malarial infection using peripheral blood that is stained and centrifuged, and examined under a UV light source. Other methods to identify malaria include fluorescence microscopy, rapid dipstick immunoassay, and Polymerase Chain Reaction. [11] In addition, testing the blood for decreased platelet levels and blood oxygenation, raised levels of bilirubin and aminotransferases may also indicate an active infection. [1] TREATMENT Malaria used to be treated with quinine-based antiparasitic drugs. This has fallen out of popularity because of the negative side effects associated with it. However, quinine is still used in some countries to treat malaria; in the United States, it is only available by prescription. [12] Chloroquine is another drug that was used to treat malaria despite its toxic side effects; however, Plasmodium species developed increasing drug resistance to it and the drug fell out of use. [13] The WHO recommends a combination of drugs known as artemisinin-based combination therapies (ACTs) for treatment of malaria. [4] Those who have been repeatedly infected by malaria may develop a partial immunity where they will have decreased severity in symptoms. [1] PREVENTION In many countries where malaria-carrying mosquitoes are prevalent, nets are used to prevent mosquito bites. [4] In addition, outdoor activity is limited during the times in which mosquitoes are most active. Insecticides such as DDT are used to prevent mosquito bites. The US has since restricted the use of DDT because of concerns for its impact on the environment; however, some mosquitoes are resistant to DDT. Some anti-malarial treatments will increase the incubation stage of the disease for as long as one year, [1] in addition, personal bug repellant sprays, repellent candles, air infusion devices are commercially available to prevent mosquito bites. REFERENCES 1. "CDC - Malaria." Centers for Disease Control and Prevention. [Online] [Accessed 17 Mar. 2010]. Available from: <http://www.cdc.gov/malaria/>. 62 2. Willey J, Sherwood L, Woolverton C. Prescott's Principles of Microbiology. 1st ed. Boston: McGrawHill Higher Education; 2009. 3. Malaria Foundation International and Emory Student Coalition Empowering Emerging Nations. "Multilateral Initiative on Malaria." Malaria Foundation International - Home. [Online] [Accessed 17 Mar. 2010]. Available from: <http://www.malaria.org/index.php?option=com_frontpage&Itemid=1>. 4. "WHO Malaria." [Online] [Accessed 17 Mar. 2010]. <http://www.who.int/mediacentre/factsheets/fs094/en/index.html>. Available from: 5. "Anemia: MedlinePlus." National Library of Medicine - National Institutes of Health. [Online] [Accessed 18 Mar. 2010]. Available from: <http://www.nlm.nih.gov/medlineplus/anemia.html>. 6. "protist." Encyclopædia Britannica. 2010. Encyclopædia Britannica. [Online] [Accessed 17 Mar. 2010] Available from: <http://www.britannica.com/EBchecked/topic/480085/protist>. 7. "Plasmodium." Encyclopædia Britannica. 2010. Encyclopædia Britannica. [Online] [Accessed 17 Mar. 2010]. Available from: <http://www.britannica.com/EBchecked/topic/463621/Plasmodium>. 8. Trampuz A, Matjaz J, Igor M, Rajesh. "Clinical Review: Severe Malaria." Critical Care Forum 7 (4): 315323. 2003. [Online -Pub MED] [Accessed 17 Mar. 2010]. Available from: <http://www.ncbi.nlm.nih.gov/pmc/articles/PMC270697/?tool=pmcentrez>. 9. "Mosquitoes, Male and Female, and Their Feeding Habits." English Vocabulary Words Mostly from Latin Greek Word Origins. [Online] [Accessed 17 Mar. 2010]. Available from: <http://www.wordsources.info/words-mod-mosquitoesPt1.html>. 10."Evolution: Library: A Mutation Story." PBS. [Online] [Accessed 18 Mar. 2010]. Available from: <http://www.pbs.org/wgbh/evolution/library/01/2/l_012_02.html>. 11. "Malaria Site: Diagnosis Of Malaria." Malaria Site: Comprehensive Malaria Website. Web. 23 Apr. 2010. <http://www.malariasite.com/malaria/DiagnosisOfMalaria.htm>. 12. "Updates--July-August 1995 FDA Consumer." Internet Archive: Wayback Machine. [Online] [Accessed 17 Mar. 2010]. Available from: <http://web.archive.org/web/20080115020839/http://www.fda.gov/fdac/departs/695_updates.html>. 13. Essentials of medical pharmacology. 5th edition; New York: -Jaypee Brothers Medical Publisher Ltd, 2003, page 739-740 63 Influenza Ndubisi Mark Chikwem General Microbiology, Spring 2010 GENERAL DESCRIPTION OF THE DISEASE Influenza (flu) is a viral disease that is caused by three strains of the influenza virus: A, B, and C. The flu has similar symptoms as the common cold and is sometimes mistaken for it. The virus contains eleven viral genes that help infect a person. The flu is more harmful than the common cold and lasts longer. Symptoms of the flu are: fever, dry cough, nausea, headache, lethargy, vomiting, and sneezing. The flu can be a pandemic or an epidemic; epidemics have killed people in the thousands and the few pandemics have killed many more. The most famous flu pandemic was the “Spanish” flu of 1918 which killed millions in only a few years. Influenza virus is part of the Orthomyxoviridae family. The virus has a negative single-stranded RNA genome that infects the respiratory tract of humans and animals. It is transmitted through coughing and sneezing which produces droplets that carry the virus, thereby facilitating its spread. Viruses can contaminate surfaces and can be transmitted through contact with the contaminated surfaces. Every year, the flu virus mutates and a new vaccine must be made. Flu viruses can also cross species to produce recombinants that are not easily prevented by a vaccine. [1- 3] SIGNIFICANCE Influenza is a seasonal disease that occurs every year, normally from November till April. This virus is airborne and therefore, spreads easily; because of this, it can lead to travel restrictions if there is an epidemic. With mutations and different strains, it is hard to treat every year. Every flu season, there is a different form of the virus that causes the flu. Due to a rearrangement of the surface proteins of the virus, it mutates and a new vaccine is made every year for the flu season. The cost to produce and distribute the flu vaccine each year is between $71 and $166 billion in America alone. The seasonal flu hospitalizes many individuals each year. Approximately every 25 years there is a flu epidemic. Unlike the common flu, a flu epidemic kills and hospitalizes people in the thousands and a pandemic kills many more. The worst pandemic was in 1918. The “Spanish” flu of 1918 killed more people more quickly than any other influenza pandemic before it. The “Spanish” flu had two major characteristics: it killed millions of people and most were between the ages of 25-40. The flu tends to be more harmful to the young and elderly because they are immune-compromised. The number of victims of the Spanish flu started off small and was not noticeable till a second and third wave hit and killed many. In one year alone, it killed 20-50 million people. The number of deaths in that one year is more than the total number of people killed by AIDS. [1] The Asian flu pandemic of 1957 killed 1-4 million worldwide in a two year span. In 1968, another pandemic called the Hong Kong flu killed one million people worldwide. There are three different strains of the influenza virus (A, B, C). Influenza A is the strain that causes pandemics. It is also the most varied of the three strains of influenza virus. Strain A has been isolated in mammalian, human, and avian species. Strain B tends to infect young children and in a few cases, adults; however, it is not as dangerous as A. Both strains B and C rarely infect humans, and are more common in animals. Not much is known about C, but it is not the cause of epidemics. [1] There are two 64 types of mutations that the influenza virus can undergo: antigenic drift and antigenic shift. Antigenic drift occurs in the hemagglutinin (HA) and neuraminidase (NA) encoding genes. [2] There are 16 subtypes of HA and 9 subtypes of NA. Different rearrangements of HA and NA subtypes produce mutations that change the virus’s genome. Mutations in the virus’s genome in the HA or NA are classified as a drift. Drift mutations are ongoing and result in a new strain variant. The new strain variant can be small or dramatic. Each year, vaccines are given out and the vaccines produce antibodies to fight against the virus. The antibodies attach to the viral HA and stop it from infecting the host cell; however, when there is an antigenic drift the antibodies cannot attach to the HA because it has mutated, and the virus can spread throughout the body. An antigenic shift is a mutation that allows the flu virus to jump from species to species. It also occurs in the HA and NA of the viral genome. There are three kinds of antigenic shifts. Shift 1 occurs with an intermediate host. When two different species pass the virus to the same animal, the two strains mix and form a new strain. Shift 2 and shift 3 do not undergo genetic changes like in shift 1; the virus can jump from species to species without changing its genome. Unlike drifts, shifts cause pandemics which cause more health problems than a drift. [2] GENERAL SYMPTOMS There are a few symptoms that doctors can use to diagnose that a patient has the flu: fever, cough, sore throat, headache, muscle aches, nausea, vomiting, and fatigue. Symptoms take 1-7 days to appear after coming in contact with the virus. It normally begins with a fever above 102˚F which usually lasts 2-5 days. Between days 2-4, the body symptoms start to subside and the respiratory symptoms increase. Most common respiratory symptoms are a dry cough; however, runny nose, headache, and sneezing may also occur. These symptoms, other than the dry cough, go away in a few days, but the cough can last for weeks. The symptoms of the flu are present for a couple weeks and then they subside. [3, 4] Influenza also leads to complications that can be used to diagnose the disease without testing. Some complications of the flu are Reye’s syndrome, Guillain-Barré syndrome and pneumonia. Reye’s syndrome damages the liver and the brain. The liver and brain go through a lot of stress, and because of that, fatty deposits are seen in the liver while edema occurs in the brain. This complication also leads to vomiting, lethargy and may result in coma. Forty percent of all cases are fatal. Taking aspirin while having influenza, is a risk factor for Reye’s syndrome. This is more common in young children because aspirin is used to control fever in children. Guillain-Barré syndrome is an auto immune disease that attacks the peripheral nervous system and can follow a viral or bacterial infection. Symptoms of this complication are weakness that can spread to the muscles of the legs and arms. This can cause the person to be almost paralyzed. It is a very rare syndrome and occurs a few days or weeks after having symptoms of a respiratory or gastrointestinal viral infection. [3, 5] Pneumonia is an inflammation of the lung tissue affecting one or both lungs. This infection usually occurs after someone has inhaled a microorganism. It can cause the person to have shivers, fever, pain in the chest and coughing. Cough starts off dry, and then it turns into coughing up phlegm, which is normally yellow and bloodstained. In severe cases, it can lead to death because the whole lung becomes inflamed. 65 PATHOGENESIS Influenza is spread easily through droplets in the air from coughing and sneezing. It invades the respiratory tract and infects the epithelial cells. Influenza virus has 11 viral genes that take part in the infection and replication of the virus: hemagglutinin (HA), neuraminidase (NA), matrix 1 (M1), matrix 2 (M2), nucleoprotein (NP), non-structural protein 1 (NSP1), non-structural protein 2 (NS2), polymerase acidic protein (PA), polymerase basic protein 1 (PB1), polymerase basic protein 2 (PB2) and polymerase basic protein 1 – F2 (PB1-F2). The virus is 80-120 nm in diameter and is spherical. The viral particle is made of a capsid containing two main glycoproteins wrapped around a central core. In the central core is the viral RNA genome. Influenza is an enveloped virus and the envelope is made up of a lipid bilayer. The virus contains two surface proteins. One of the surface proteins (HA) binds the virus to the host cell and the other (NA) allows the virus to leave the host cell. HA forms spikes on the lipid membrane of the virus. These spikes bind to the sialic acid on the membrane of the host cell. HA starts the entry into the host cell. After binding to the host cells’ sialic acid, receptor mediated endocytosis occurs, and virus enters the cell through an endosome. As virus enters the endosome, fusion occurs between the viral and endosomal membrane. The endosome has an acidic pH which opens up the M2 ion channel. The opening of the M2 ion channel uncoats the virus and releases the viral RNA which then enters the cytoplasm. [9] To infect the cell, the vRNA must enter the nucleus. The viral proteins NP, PA, PB1, and PB2 facilitate the entry of the vRNA into the nucleus. These proteins locate and get the vRNA into the host cell’s nucleus. After entering the nucleus, the negative strand viral RNA must be converted to positive strand RNA that is used as a template to produce more viral RNA. RNA dependent RNA polymerase (RdRp) starts RNA synthesis internally on the viral RNA template. The new viral RNA either stays in the nucleus or is moved to the cytoplasm. The viral RNA that is moved to the cytoplasm is translated into proteins. The synthesized viral proteins are either secreted through the Golgi apparatus onto the cell surface or back into the nucleus to bind to viral RNA and form genomic particles. [9] Viral RNA encodes new HA and NA that cover the cell membrane of the host cell. The mature virus leaves the host cell by budding, and uses the host cell's cell membrane to re-envelope itself. On the cell membrane is the HA and NA which the virus needs to leave the cell and infect new ones. After budding and before being completely free from the host cell, the virus must cleave the sialic acid. NA cleaves the sialic acid and frees the virus from the host cell. This is the most important step, because without the cleavage of the sialic acid, the virus will not be able to leave the host cell and infect other cells. This then results in the host having less severe and shorter symptoms of the flu. [9] In summary, the mechanism of entry and replication of the virus is well known; but, the pathogenesis of influenza has not been clearly elucidated. Some new studies, however, suggest that apoptosis of alveolar epithelial cells may play a major role. One study concludes that the viral destruction of alveolar epithelial cells results in pneumonia and the destruction of leukocytes, leading to leucopenia which is a prominent clinical feature of influenza in humans. [10] 66 LABORATORY DIAGNOSIS During epidemics, a diagnosis of the flu can be made on the basis of clinical symptoms alone. However, influenza A and B strains can co-circulate and cause mixed infections of influenza with other viruses, so, laboratory diagnosis must be done to specify which strain is present. Different techniques can be used in the lab to make a diagnosis: virus isolation, rapid diagnosis by immunofluorescence, and serology. Virus isolation is done with throat swabs, but nasal washings are the best samples. The sample can be inoculated in either embryonated eggs or tissue cultures. Embryonated eggs that are 10-12 days are inoculated into the amniotic cavity. The virus then replicates in the amniotic membrane and large amounts of the virus are released back into the amniotic fluid. After 2-3 days of incubation, aliquots of harvested amniotic fluid can be used for viral detection and hemagglutination test using chick, guinea pig, or human erythrocytes. [7] Newly produced viruses can also be recognized by haemadsorption using the cells in the tissue culture. Also haemagglutination can be performed using the culture medium which contains free virus particles. The viruses isolated from the embryonated eggs or tissue culture can be identified by serological or molecular methods. The strains can be recognized using a complement fixation test against the soluble antigen. Rapid diagnosis by immunofluorescence tests cells from pathological specimens for the presence of antigens made by stains A and B. To detect the antibodies in body fluids, the antibody probes are labeled with a fluorescent dye that produces green color when viewed with a microscope under ultraviolet light. Serology is used when the virus cannot be isolated. This is a blood test that detects the presence of antibodies against a specific microorganism. Because the body produces specific antibodies which target certain microorganisms during an infection, the blood can be checked for those specific antibodies, and if no antibodies are present for the microorganism you are looking for, then that microbe usually is not present in the body. [7] TREATMENT Antiviral drugs are used to treat influenza; however, they are not a cure for the flu. They do not get rid of the symptoms, but they decrease the severity of the symptoms, and the duration of the flu by 2-3 days. Antiviral drugs include: oseltamivir, zanamivir, rimantadine, and amantadine. The drug to take will depend on the strain of the virus. Neuraminidase inhibitors are antiviral drugs that stop the spread of the influenza virus. Neuraminidase inhibitors are analogues of sialic acid and they stop the virus from releasing from the host cell after budding has occurred. The analogues cap the NA on the viral cell and stop it from cleaving the sialic acid that the HA is bound to. There are two main Neuraminidase inhibitors used today: oseltamivir and zanamivir. They are both highly effective against influenza A and B, but they must be taken 1-2 days after first contact to stop the spread of the virus. After 1-2 days, the virus has spread to too many cells in the body for the drug to have a significant effect. Zanamivir is given as a dry powder and must be applied topically to the cells in the respiratory system via inhalation. It has a low oral bioavailability and because of this, another inhibitor, Oseltamivir is recommended. Oseltamivir is taken orally and has a high bioavailability. At the moment, some strains of the virus are resistant to oseltamivir in the United States. [3, 6] 67 Rimantadine and amantadine are drugs that stop the virus from entering the cell. They block the virus from entering the endosome, and they also stop the release of the virus from the cell. These drugs work well against strain A, but not B. Rimantadine and amantadine are not being used at the moment because of resistance. All antiviral drugs must be taken no longer than two days after symptoms have occurred, or other forms of treatment should be used. [6] Bed rest is also needed to recover from the flu. PREVENTION One method of prevention is the administration of flu vaccines. Vaccines are used for people at high risk of infection; they have the greatest effect on the seasonal flu, but not epidemics. In the case of the flu, the young and the elderly are at highest risk for infection. Due to their undeveloped and/or weak immune system, they cannot fight off the infection like young, healthy adults. Influenza vaccines now carry three virus strains: two types of strain A and one type of strain B. The three strains in the vaccine represent the three types of the viruses predicted for the upcoming flu season. Because the virus mutates every year, the vaccine is not the same every year. The vaccine is made from inactivated viruses grown in eggs. People who have taken the vaccine develop haemagglutination-inhibition antibody titers. These antibodies protect them from illness caused by the strain of the virus in the vaccine. Elderly people may produce fewer antibodies than young, healthy adults, and therefore, can still develop upper respiratory tract infections when they come in contact with the influenza virus, but the infection will not need hospitalization and will not lead to death. [7] The vaccine must be taken a few weeks before coming in contact with the virus to have an effect. Time is necessary for enough antibodies to be produced. There are two types of vaccines: trivalent inactivated influenza vaccine (TIV) and live-attenuated influenza vaccine (LAIV). TIV is an inactivated vaccine that contains viral proteins but has no live virus. It is administered through an injection, and can be given to people six months and older. Some symptoms that might occur after injection are soreness at injection site, fever, nausea, lethargy, headache, muscle aches, and chills. These are the same symptoms that are seen in patients infected with the flu. Because the vaccine has the viral proteins, the body detects the foreign proteins and begins an immune response to get rid of it. This may cause the symptoms seen in an actual infection, but the patient is not infected or ill. LAIV is administered nasally and can be given to people ages two and up. LAIV also has the potential to have the same symptoms as the flu. Both TIV and LAIV are grown in eggs and cannot be administered to people who have allergic reactions to chicken and egg proteins. Other ways to prevent the infection is to wash ones hands and to stay away from people who have symptoms of the flu. [7] During epidemics, infected people should be quarantined, and those showing symptoms but not severely ill should be advised to stay away from work. REFERENCES 1. White J. Medical Ecology. [homepage on the Internet]. 2005 [cited 2010 Mar 14]. Available from: http://www.medicalecology.org/diseases/influenza/influenza.htm#sect2.1 68 2. Pike J. Homeland Security: Antigenic Drift vs Antigenic Shift. [homepage on the Internet]. 2009 [cited 2010 Mar 14]. Available from: http://www.globalsecurity.org/security/ops/hsc-scen-3_flu-antigenic.htm 3. Hunt M. Microbiology and Immunology: Influenza. [homepage on the Internet]. 2009 [cited 2010 Mar 14]. Available from: University of South Carolina School of Medicine, Web site: http://pathmicro.med.sc.edu/mhunt/flu.htm 4. Medicinenet: Influenza. [homepage on the Internet]. 2009 [cited 2010 Mar 14]. Available from: Centers for Disease Control Web site: http://www.medicinenet.com/influenza/page2.htm#symptoms 5. NINDS Guillain-Barré Syndrome. [homepage on the Internet]. 2009 [cited 2010 Mar 14]. Available from: National Institutes of Health Web site: http://www.ninds.nih.gov/disorders/gbs/gbs.htm 6. Leblebicioglu H, Brook I. Influenza: Treatment & Medication. [homepage on the Internet]. 2009 [cited 2010 Mar 14]. Available from: Web site: http://emedicine.medscape.com/article/972269-treatment 7. AMERICAN ACADEMY OF PEDIATRICS. [serial on the Internet]. 2009 [cited 2010 Mar 14]. Available from: http://aapredbook.aappublications.org/news/FluPolicy2009-10.pdf 8. Laboratory Diagnosis of Influenza Viruses Infection. [homepage on the Internet]. 2010 [cited 2010 Mar 14]. Available from: http://virology-online.com/viruses/Influenza5.htm 9. Samji T, Influenza A: Understanding the Viral Life Cycle. YALE JOURNAL OF BIOLOGY AND MEDICINE 2009; 82:153-159. 10. Uiprasertkul M, Kitphati R, Puthavathana P, et al. Apoptosis and pathogenesis of avian influenza A (H5N1) virus in humans. Emerging Infectious Diseases 2007; Vol 13 (5): 708 - 712 69 Clostridium botulinum Stacey-ann Wright General Microbiology, 2010 DESCRIPTION OF THE AGENT Clostridium botulinum is a rod-shaped, Gram-positive bacillus that is typically arranged in singles, pairs or chains. C. botulinum can remain dormant due to the formation of spores until the right environmental conditions are present. Its spores are very resistant to extreme environmental conditions such as heat, radiation and chemicals, which makes them very hard to kill. Favorable growth conditions include anaerobic, acidic and substrate-rich environments. C. botulinum also prefers to grow in media with salt level lower than 7%, sugar content lower than 50%, temperatures between 400F and 1200F (4-490C), and high moisture content. [1] The genome size of Clostridium botulinum is estimated to be around 4039kbp. [2] The genome size is relatively larger than many other Gram-positive organisms. This could possibly indicate that extra genomic requirements are needed for sporulation and disease-inducing toxins. SIGNIFICANCE This organism was discovered and identified by Emile Van Ermengem in 1896. [2] This organism is of interest because it possesses one of the most potent toxins known to man. It is the most acute toxic substance known, with a median lethal dose of about 1 ng/kg when introduced intravenously, [3] and 3 ng/kg, when inhaled. [3] This means that, depending on the method of introduction into the body, 90– 270 nanograms of botulinum toxin could be enough to kill an average size person. The importance of sequencing the genome of C. botulinum lies in its ability to produce a deadly toxin known as botulin, this powerful toxin leads to the paralytic illness known as botulism. Clostridium bacteria are widely distributed throughout the world. Clostridium botulinum occurs as both vegetative bacterium and spores in soils, in marine sediments and on the surfaces of fruits. In Europe, contaminated hams and sausages are the most common modes of transmission. Poland has the highest incidence of botulism by far, with 325 outbreaks and 448 cases in a 3-year period. China is a distant second with 38 outbreaks and 168 cases in a 25-year period. [4] Most countries have not yet reported a case of infant botulism, most likely because of not recognizing or not reporting or both. Before 1900, the mortality rate associated with botulism was 70%. Today, the mortality rate approaches 15%. [4] Almost any food with a pH of 4.6 or higher can support the growth of the bacterium and the subsequent production of toxin. Humans (both adults and infants) regularly ingest the spores, but rarely suffer ill effects unless the immune system is compromised. This is because the immune system destroys the spores before they can germinate and produce toxin. C. botulinum causes three main categories of botulism namely; food-borne botulism, wound botulism, and infant botulism. Food-borne pathogenic bacteria are responsible for one of the most expensive healthcare cost to the government and food industry. Organisms in this category include Staphylococcus, Escherichia coli, Salmonella and C.botilunum. There are 10-30 outbreaks (between 25110 individual cases) of botulism reported in the United States each year. Approximately 75-100 70 individual cases are infant botulism. [1] Infant botulism is of concern as C. botulinum spores have been found in honey that was implicated in infant botulism. The CDC reports that of the 145 cases of botulism occurring per year, food-borne, infant, and wound botulism constitute 15%, 65%, and 20% of cases, respectively. [5] In 2010, a new study by the FDA showed that the total economic impact of food-borne illness across the nation is about 152 billion dollars annually. [5] C.botulinum is not only significant due to the diseases it causes, but also as a bioweapon where terrorists attempt to aerosolize the bacterial toxin. If inhaled in the aerosolized form, a single gram of crystalline toxin, evenly dispersed and inhaled, would kill more than 1 million people, although technical factors would make such dissemination difficult. [6] Although botulinum toxin is a lethal naturally occurring substance, when carefully isolated and purified, it can be used as an effective and powerful medication. Researchers discovered in the 1950s that injecting overactive muscles with minute quantities of botulinum toxin type-A would result in decreased muscle activity by blocking the release of acetylcholine from the neuron, thereby preventing the vesicle where the acetylcholine is stored from binding to the membrane where the neurotransmitter can be released. This will render the muscle unable to contract for up to a period of three to four months. In cosmetics, a Botox injection, consisting of a small dose of the botulinum toxin, can be used to prevent the formation of wrinkles by paralyzing facial muscles. As of 2007, it is the most common cosmetic operation, with 4.6 million procedures in the United States. [7] The wrinkle preventing effect of Botox lasts for approximately 3-4 months, up to 6 months. [7] In addition to its cosmetic applications, Botox is currently used in the treatment cervical dystonia (a neuromuscular disorder involving the head and neck), excessive blinking, excessive sweating and migraine. Other uses of botulinum toxin type A that are widely known but not specifically approved by FDA, include treatment of diabetic neuropathy, wound healing and excessive salivation. [8] PATHOGENESIS Clostridium botulinum can enter any adult's body; however, natural defenses would prevent the spore from germinating, but if these natural defenses are compromised, the spores will germinate. Humans can come into contact with botulin by eating improperly canned or preserved foods that contain the botulinum toxin, having an infected wound or by inhaling the bacterium in its pure form. The spores of Clostridium botulinum are not in themselves dangerous but the toxin secreted is of great concern. This is produced only when the organism is actively metabolizing i.e. the right conditions have been provided. During growth, Clostridium botulinum produces at least seven different toxins such as neurotoxins, enterotoxins and haemotoxins, which include some of the most potent toxins ever discovered. In rare cases, one strain may produce more than one type of toxin. Once it gets into the body, Botulinum toxin predominantly affects the peripheral nervous system (PNS) specifically ganglionic synapses, postganglionic parasympathetic synapses, and myoneural junctions (the nerve endings where the nerves join muscles and where the toxins block motor nerve terminals). In the body, neurotransmitters are the chemical messengers used by nerve cells to communicate with each other and also with muscles. The botulism toxin causes the characteristic flaccidity of muscles by preventing the release of acetylcholine. 71 SYMPTOMS Characteristic symptoms of an infection with Clostridium botulinum include abdominal pain, vomiting, motor disturbances and difficulty seeing. Symptoms of food-borne botulism usually occur 12-36 hours after the ingestion of food contaminated with C. botulinum; however, this can take 10 days to manifest. [1] Early symptoms include general weakness, vertigo and dry mouth. Symptoms that follow are usually more intense which include blurred and double vision, drooping eyelids, progressive difficulty in speaking and swallowing, a nasal or hoarse voice, difficulty in breathing, muscle weakness, abdominal distension and constipation. Nausea and vomiting are normal reactions that result from swallowing the organism. Infants who are infected with C. botulinum first present with constipation which may go unnoticed by parents, as most infants get constipated very often. Infants then become lethargic and floppy, unable to hold their heads up. Oral secretions may be accumulated. Infants are also unable to form expression, crying will become altered until they cannot cry anymore, and they will not be able to feed or suck because of the nerves that this toxin affects. Infants may display symptoms 3-30 days after ingestion. [1] The symptoms of wound botulism are the same as food borne botulism. Symptoms occur 4-14 days after exposure. [1] This is the only type of botulism where antibiotics can be used. DIAGNOSIS For infant botulism, diagnosis should be made on clinical grounds. Confirmation of the diagnosis is made by testing the stool or enema specimen with a mouse bioassay. Patient history and physical examination may be considered; however, these clues are often not enough to allow a diagnosis. Special tests may be needed to rule out other diseases that are similar to the symptoms of C. botulinum infection such as Guillain-Barré syndrome. These tests may include a brain scan, cerebrospinal fluid examination, nerve conduction test, and an endrophonium chloride test for myasthenia gravis. A definite diagnosis can be made if botulinum toxin is identified in the stomach or intestinal contents, vomit or feces. Botulinum toxin can be detected by a variety of techniques including enzyme linked immunobsobent assays (ELISA), electrochemiluminiscent test (ECL), and mouse inoculation or feeding trials. In infectious botulism, the organism can be cultured from tissues on Blood agar medium or egg yolk medium. Toxin producing colonies usually display surface iridescence that extends beyond the colony. The most direct way to confirm the diagnosis of botulism in the patient’s serum or stool is by injecting serum or stool into mice as mentioned above. The commercially available ELISA for botulinum toxin antibodies is not recommended because it has significant false-positive rates and has not been studied in evaluating patients with botulism. PREVENTION Prevention of botulism is based on good food preparation (particularly preservation) practices and hygiene. One possible way of destroying the botulinum toxin is by cooking over the course of a few minutes; the spore itself is not killed by the high temperatures; however, leaving the cooked material overnight may induce the spore to germinate and produce the toxin. For infant botulism, the only possibly way of preventing infection is to avoid feeding honey or corn syrup to infants less than 12 months of age, and to monitor how feed is stored. For food borne botulism, canned food is a major way in which this organism can be passed on. Botulism can be prevented by pressure cooking foods at 250 F (121 C) for at least 30 minutes. [9] Also, canned foods should be boiled for 10 minutes before serving 72 them. Other prevention techniques include not storing herbs or garlic covered in oil that has not been acidified, improperly handling potatoes wrapped in aluminum foil and improper home canning and fermenting fish. If home canning is done, home canned foods should be boiled for 20 minutes. Metal cans and containers in which bacteria are growing may bulge outwards due to gas production from bacterial growth; such cans should be discarded immediately. Wound Botulism can be prevented by immediately seeking medical care for infected wounds and by avoiding punctures with unsterile things such as needles used for street drug injections. If exposure to the toxin via an aerosol is suspected, the clothing of the patient must be removed and stored in plastic bags until it can be washed with soap and water. This will help to prevent additional exposure to the patient and health care workers. The patient must shower thoroughly. Food and water samples associated with suspected cases must be obtained immediately, stored in proper sealed containers, and sent to reference laboratories for analysis and diagnosis. TREATMENT In the case of food-borne botulism, doctors may clear the digestive system by inducing vomiting and giving medications to induce bowel movements. If the patient has wound botulism, the doctor may have to remove the tissue surgically and treat for symptoms. If the individual is diagnosed early with wound or food-borne botulism, equine antitoxin that is injected reduces the chances of complications and for wound botulism alone, the patient may be given antibiotics such as penicillin. Anti-toxin attaches itself to the toxin that is still circulating in the bloodstream, and keeps it from affecting nerves. Anti-toxin is not recommended for infants since it doesn’t affect the disease causing germs in the baby’s digestive system. Therefore for infants, if the symptoms are recognized early, they can be treated and supported with tube feeding and respirators. A treatment called botulism immune globulin has been investigated to treat infants. It appears effective in reducing the duration and severity of the illness. Problem with breathing is associated with botulism infection so the patient may need a mechanical ventilator to force air into the lungs. Patient may remain on ventilator for weeks as toxin gradually lessens. There is a vaccine against botulism, but it is used very rarely as its effectiveness is not fully evaluated and it has side effects. REFERENCES 1.Harper TK. Clostridium botulinum. Tara Harper [Homepage on internet . [updated 2010 Feb 02] [cited 2010 Mar 14] .Available from: http://www.tarakharper.com/b_botuln.htm#des. 2. Peralta, Brannon. Microbe Wiki. Clostridium botulinum . [Homepage on internet]. [updated 2007 July 13] [ cited March 16, 2010 ]. Available from: http://microbewiki.kenyon.edu/index.php/Clostridium_botulinum . 3. Gill DM. Bacterial toxins: a table of lethal amounts. Microbiol Rev. 1982; 46: 86 – 94. 73 4. Wrong diagnosis. Statistics by country of botulism food poisoning. [Online] [cited March 18, 2010]. Available from: http://www.wrongdiagnosis.com/b/botulism_food_poisoning/stats -country.htm 5. Pew Health Group. Foodborne Illness Costs US $152 Billion Annually Landmark Report Estimates. [Homepage on internet] [updated 2010. March]; [cited April 2, 2010] Available from: <http://www.sciencedaily.com/releases/2010/03/100303081834.htm>. 6. Arnon, SS. et al., "Botulinum Toxin as a Biological Weapon". JAMA. 2001; 285: 1059–1070. [Online] [Cited April 2, 2010]. Available from http://www.cosmeticplasticsurgerystatistics.com/statistics.html 7. Hallett M. One man’s poison – clinical applications of botulism toxin. N Engl J Med. 1999; 341 (2): 118 – 120 8. Cardosso F, Jancovic J. Clinincal use of botulinum neurotoxins. Curr Top Microbiol Immunol. 1995; 195: 123 – 141 9. [Guideline] Bossi P, Tegnell A, Baka A et al. Bichat guidleines for the clinical management of botulism and bioterrorism-related botulism. Euro. Surveill. 2004; 9: E13 -14 [Medline] 74 HIV: Human Immunodeficiency Virus Stephanie Rand General Microbiology, Spring 2010 DESCRIPTION Human Immunodeficiency Virus, or HIV, is a parasitic retrovirus, which infects and destroys human CD4+ and T4 cells. In doing so, HIV suppresses the body’s immune system, leaving the host susceptible to opportunistic infections. The last stage of HIV, which is characterized by opportunistic infections, is known as AIDS, Acquired Immunodeficiency Syndrome. HIV is an animal lentivirus, typically 100-150 nm in length. The HIV virion is an enveloped virus, covered with 72 external spikes, or peplomers. [1] Each peplomer is composed of the glycoproteins gp120 and gp41 (gp160 env gene product). Gp41 extends through the membrane, and is a coiled protein. Gp120 is attached to gp41, and extends beyond the membrane. Together, gp160 plays a vital role in the virus’s binding to the host cell. The virus’s envelope is also a reflection of the CD4+ cell’s histocompatibility complex, acquired by the virus via budding from the cell. Within the lipid bilayer, the nucleocapsid contains two identical strands of single stranded HIV RNA, and the enzymes Reverse Transcriptase (p51/66), Protease (p10) and Integrase (p31). [2] Within HIV’s genome are nine recognizable genes, producing 15 different proteins. Proteins are divided into three categories: structural, regulatory and accessory. Structural proteins include GAG, POL and ENV. GAG, or group-specific antigen, has been dubbed the “virus particle-making machine” as it both codes for internal protein structures, and gives HIV the faculty to bud from the host cell. POL gene codes for the enzymes (reverse transcriptase, integrase and protease) which are contained within HIV. ENV codes for gp 120 and gp41 (surface proteins). Both regulatory and accessory proteins collectively control HIV reproduction. Regulatory proteins include Tat and Rev. The tat gene acts as a transcription activator. The rev gene regulates the viral protein expression by splicing RNA and moving it to the cytoplasm. Accessory proteins are Nef, Vif and Vpu. Nef, or negative effector, is believed to protect the cell from premature death, so that it may be used to produce more HIV virions. Vif, viral infectivity protein, is believed to neutralize the human enzyme APOBEC, which has the potential to stop HIV production. Lastly, Vpu codes for viral protein U. Viral protein U is responsible for wiping out CD4 proteins, increasing the ease of HIV assembly and subsequent budding from the cell. [3] SIGNIFICANCE HIV is a pandemic that infects persons of all ages, races, socioeconomic status, nationality and sexual orientation. Once the virus has been integrated into the host cell’s genome, infection is irreversible. There is no viable cure for the virus, although treatments which extend the asymptomatic period are available. It is estimated that HIV has led to 25 million deaths worldwide since 1981. In 2008, approximately 2.7 million people were newly infected with HIV; estimates for total infected persons worldwide are roughly 33 million people. [4] In the United States, it is estimated that 1,106,400 persons are living with HIV as of 2006. [5] While HIV infects millions, it also affects countless others. Approximately 15 million children under the age of 18 have been orphaned by AIDS worldwide. [6] 75 Losing even one parent or family member can have serious consequences on the household provisions, education and family structures. Those who are infected with HIV are often stricken in the prime of their high yielding work potential years. Illness associated with HIV infection decreases the infected person’s productivity and ability to provide economically and otherwise for their families. Communities with exceptionally high infection rates, have entire population segments which are out of work and unable to provide and care for their families. HIV is not only a physically detrimental infection, but economically disadvantageous as well. Antiretrovirals cost an average of $2,100 a month, leading to an average lifetime cost of $618,900 for an infected person. [7] As many infected persons cannot cover these costs, they are often absorbed by government programs. In addition, governments funnel millions of dollars into prevention programs and support services to HIV positive individuals. GENERAL SYMPTOMS After initial HIV infection, symptoms occur in four phases: acute infection, asymptomatic phase, chronic symptomatic phase, and lastly, AIDS. The first stage, initial infection, spans the first year after the virus has invaded the body. Approximately 70% of infected individuals will experience flu-like symptoms (fever, headache, swollen lymph nodes, sore throat, nauseas, vomiting, rash diarrhea, thrush, etc.) about 2 to 4 weeks after initial infection. However, as these symptoms will generally persist for only one to four weeks, they are typically written off as a common virus. During the initial weeks and months post infection, the virus is rapidly duplicating itself and infecting immune system cells, including monocytes, macrophages, T4/CD4+ cells and follicular dendridic cells. While copying of the HIV virion may be as high as 10 million/ml of blood, antibody production by the immune system is fairly low. Labeled the window period, or window of infectivity before seroconversion, this 3 to 6 week sub-phase is characterized by high viral growth and little immune response. Because most HIV testing methods test for HIV antibodies, a test on an HIV positive individual in their window period will not be positive. Because viral load is so high and unchecked at this point, the host is highly infectious. The asymptomatic stage of HIV infection lasts approximately 10 years. While an HIV infected person in the asymptomatic phase exhibits no symptoms, the virus continues to multiply and destroy T4 and CD4+ cells, while the body attempts to produce HIV antibodies and replace destroyed T4 cells. In spite of showing no symptoms, individuals are still highly infective. The symptomatic stage of HIV infection can be broken down into two divisions: Early HIV-related symptoms and advanced HIV-related symptoms. Early symptoms include immune thrombocytopenic purpura, mild anemia, mild leukopenia, and a decrease in T-cell count. Advanced symptoms include moderate anemia/leukopenia, low albumin/cholesterol and further T cell decrease. As the immune system becomes overwhelmed by an increase in viral load and decrease in T-cell count, other general symptoms include: headaches, weight loss, fever, malaise, fatigue, stomach pain, night sweats, diarrhea, etc. While normal T cell count is about 1000+ per/μL, during this stage the count decreases. At 500 or less T cells per μL of blood, fungal and bacterial infections such as thrush or oral lesions are common. 76 The last stage of HIV infection, AIDS, is a diagnosis made by a physician when either T cell count drops below 200 cells/μL of blood, or opportunistic infections are present. Opportunistic infections are viral, bacterial, fungal or protozoan infections that, while normally held in check by a healthy immune system, can easily overcome an immune system which has been weakened by HIV infection. Viral infections include Hepatitis C, and Cytomegalovirus. Common protozoal diseases are Toxoplasma gondii and Cryptosporidium. Common fungal opportunistic infections include Histoplasmosis, Candidiasis/Thrush and Pneumocystis carinii (PCP now referred to as Pneumocystis jiroveci). Myobacterium avium, M. intracellulare and Mycobacterium tuberculosis are common bacterial opportunistic infections. Some cancers qualify as AIDS-defining illnesses. Kaposi’s sarcoma, characterized by skin lesions, is a common cancer in infected men, and is possibly caused by the Human Herpes Virus. AIDS Related Lymphomas (ARL), Non-Hodgkin’s Lymphoma, Progressive Multifocal Leucoencephalopathy and invasive cervical cancer are other AIDS-defining cancers. [3] PATHOGENESIS HIV spreads from human to human via blood, breast milk and vaginal or seminal fluids from infected persons. This can occur via vaginal, anal or oral sex with an infected person, IV drug use and blood transfusion (or other needle use that transfers blood from infected person, i.e. tattooing), and from an infected mother to her child via breast milk, or blood transfer during pregnancy or childbirth. [6] Once HIV is able to enter the bloodstream of the host, it begins to infect monocytes, macrophages, T4/CD4+ cells and follicular dendridic cells. [3] HIV’s primary target is the CD4+ cell. Gp120 and gp41 play an important role as co-receptors in the fusion of the HIV to the CD4+ membrane. While collision is random, the gp160 spike is specific to R4 and R5 receptors on the CD4+ membrane. R4, CXCKR4 chemokine receptor, is found on CD4+ lymphocytes or T4 cells. Also a chemokine receptor, R5, CCKR5 receptors are found on macrophages and CD4+ cells. When the HIV virus fuses to the CD4+ membrane via the receptors, the coiled gp41 is “un-sprung” into the host cell membrane. The nucleic acid is then uncoated, and the two strands of identical single stranded RNA are released into the cytoplasm. Because HIV is a retrovirus, it must transcribe dsDNA from its ssRNA, which can then be integrated into the host cell genome. HIV accomplishes this via the enzyme reverse transcriptase. Reverse transcriptase first uses its polymerase portion to make a complementary strand to the ssRNA. Then, the RNase region of the enzyme detaches the ssRNA from the complimentary ssDNA. The polymerase portion is again used to create a complimentary DNA strand, resulting in dsDNA of HIV, which can be integrated into the host cell genome via the enzyme integrase. Once the HIV dsDNA has been integrated into the host cell genome, the CD4+ cell is deemed a provirus. Once the cell has become a provirus, infection is irreversible. That is, every time the CD4+ cell is replicated, it will also replicate the HIV dsDNA along with its own. Once the CD4+ cell has become a provirus, transcription occurs in order to produce viral components. HIV harnesses the cells own transcription mechanisms in order for mRNA to undergo transcription. This process cannot begin until the long terminal repeats at the ends of the provirus turn on the RNA polymerase. The RNA produced by translation of the provirus is either mRNA, which is transcribed into 77 proteins for viral components, or ssRNA to be enclosed within the new viruses as their genome. The production of the two respective forms of RNA occurs in two distinct phases. The first phase of translation of RNA (early phase) is used as mRNA. Approximately 2000 nucleotides in length, the mRNA is translated into regulatory proteins after being spliced by cellular enzymes located in the cytoplasm. The secondary phase of provirus translation (late phase) produces both the RNA which will compose the new viruses genome, as well as nucleotides which code for the enzymes and structural proteins for the new HIV virions. Once the regulatory proteins, genomic ssRNA, and structural and enzymatic proteins have been synthesized by the host cell, the components must come together in order to escape the cell and infect other CD4+ or T4 cells. In order to bud out of the cell, HIV harnesses the CD4+ cells standard system. After HIV infects the T4 or CD4+ lymphocyte, it has an approximate life span of 48 hours. Each new generation has the potential to mutate, changing the genetic makeup of the virus. Unlike the typical animal cell, the process of initial transcription of the viral dsDNA from ssRNA lacks any proofreading mechanism. In transcription of dsDNA, DNA polymerase III is a 10 protein composite, where 2 of the 10 proteins are core enzymes. In typical transcription of DNA these core enzymes act as proofreading mechanisms. [1] On the contrary, RNA viruses have a much higher mutation rate, and lack the proofreading mechanism afforded by the presence of DNA polymerase III. Furthermore, reverse transcriptase has a particularly high error rate. Both the rate of mutation, and lack of proofreading accounts for the high genetic diversity of the virus. This enhances HIV’s ability to develop drug resistance and evade the body’s immune responses. [3] LABORATORY DIAGNOSIS Diagnosis of HIV typically occurs through an HIV antibody test. HIV antibody tests detect HIV antibodies in a person’s body fluids. [2] HIV tests either detect the antibody produced by the immune system in response to infection, actually identifies the HIV antigen, detects the viral specific genetic material, or provides a T cell count. Those tests that detect antibodies are referred to as indirect tests, as they only suggest the presence of HIV. Direct tests, on the other hand, are those which specifically detect the nucleic acid components of HIV. While all tests can be used, antibody testing is the most common and affordable method. Tests include ELISA (enzyme linked immunosorbent assay), Aplicor’s branched DNA test, western blot, immunofluorescent antibody assay, PCR (polymerase chain reaction), and rapid HIV kits. TREATMENT While there still exists no effective cure for HIV infection, 24 individual drugs and 6 combination drugs have been developed and approved by the FDA for use in combating HIV infection. Although available drugs are effective in keeping the virus in check, they all are accompanied by harmful and unpleasant side effects. Antiretroviral drugs are divided into several drug classes: nucleoside analog reverse transcriptase inhibitors, HIV-protease inhibitors, non-nucleoside reverse transcriptase inhibitors, entry inhibitors, integrase inhibitors, maturation inhibitors (currently under investigation), and AV-HALTs. 78 Nucleoside reverse transcriptase inhibitors (NRTIs), or nukes, are incorporated into viral DNA, preventing HIV replication by inducing elongation of viral DNA which leads to chain termination. Reverse transcriptase is unable to join the next nucleoside. NRTIs include Zidovudine, Didanosine, Zalcitabine, Stavudine, Lamivudine, Combivir, Abacavir, Trizivir, Tenofovir, Emtricitabine, Truvada, Epzicom, and Atripla. Many NRTIs are accompanied with extreme side effects, including lethargy, headache, inhibition of the replication of mitochondrial DNA, pancreatitis, anemia, etc. HIV-protease inhibitors bind competitively to the active site on HIV-protease. This prevents cleavage of HIV proteins. The resulting particles are noninfectious. Protease inhibitors include Saquinavir mesylate, Ritonavir, Indinavir, Nelfinavir, Saquinavir, Amprenavir, Keletra, Atazanavir, Fosamprenavir, Tipranavir and Darunavir. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), or non-nukes are structurally and chemically dissimilar groups of antiretrovirals that can be used effectively in triple-therapy regimens. [2] NNRTIs undergo non-competitive inhibition of reverse transcriptase in order to hinder the replication of HIV. NNRTIs include Nevirapene, Delaviradine, and Efavireuz. The goal of NNRTI use is to extend the patients asymptomatic period, while postponing initiation of protease inhibitors. NNRTI should be used in combination therapy with other drugs, as NNRTI resistant mutants arise quickly in the presence of monotherapy. Entry/fusion inhibitors inhibit HIV’s ability to bind with and enter the host cell. Entry/fusion inhibitors include T-20, Maraviroc, FP21399, PRO 542 and 140, TNX-355, etc. Integrase inhibitors prevent the integration of HIV into host cell DNA, precluding formation of a provirus. Raltegravir is an integrase inhibitor. The goal of HIV treatment is two-fold: lower viral load and increase CD4+ or T4 cell count. Although monotherapy of AZT was used in the late 1980s and early 1990s, it proved to be ineffective in inhibiting HIV production in the long term. When a single drug is taken by an HIV positive person, viral load will decrease as drug sensitive HIV is prevented from reproducing. However, due to HIV’s high rate of mutation, drug-resistant mutants that have developed will continue reproducing, eventually resulting in a drug resistant strain of HIV. Combination therapy, or HAART (Highly Active Anti-Retroviral Therapy), acts by suppressing viral load through the combination of multiple antiretrovirals. HAART decreases the development of drug resistant strains of HIV, as a successfully drug resistant mutant would have to develop multiple resistances in response to each drug. The greater the number of genetic changes that must occur, the lower the likelihood of a resistant strain forming. In some cases, HAART therapy fails to effectively control viral load. Regimen failure can occur due to virologic failure, immunologic failure, or clinical failure. Virologic failure occurs when HIV does not become undetectable after over two years of treatment. Immunologic failure occurs when CD4+ count falls below the levels measured prior to therapy initiation. Clinical failure occurs if there is still a decrease in physical health. When HAART failure occurs patients must resort to salvage therapy/mega 79 therapy/giga HAART therapy. Salvage therapy is defined by therapy following at least three failed drug regimens (one from each drug class). [3] PREVENTION As HIV has several modes of transmission, prevention methods are numerous. Although abstinence is the only 100% effective method of preventing sexual transmission of HIV, barrier methods, such as a latex or polyurethane female or male condom or dental dam, are effective measures in preventing transmission through anal, oral or vaginal sex. Being monogamous in a relationship also significantly reduces risk of HIV transmission. HIV transmission in intravenous drug users can be prevented through exclusively using new, sterilized syringes. Needle exchange programs provide new, sterile syringes in exchange for a used one. Needle exchange centers also generally provide sterile drug paraphernalia to and counseling/risk reduction programs for drug users. Other blood to blood transmission is virtually impossible in the United States due to rigorous blood screening, including a minimum of 52 medical restrictions for blood donation. However, transmission statistics in developing countries point to blood transfusions as the cause of 10% of new HIV infections. [3] Mother to child transmission can occur during pregnancy, childbirth, or through breastfeeding. Universal HIV testing of pregnant women is an important component in preventing mother to child transmission, as many pregnant women may not be aware of their HIV status. A lower viral load in the mother will translate to a lower transmission risk to the child. Thus, antiretrovirals taken by the HIV positive mother can reduce transmission risk. In addition, a single dose of Nevirapine halves the risk of transmission, and is often the method used in developing countries. However, approximately 1/3 of women treated with one dose of Nevirapine will develop drug resistant HIV, and half of children born HIV positive will be infected with drug resistant HIV. During childbirth, a cesarean section can reduce the risk of transmission to the child due to trauma in the birth canal. Mothers are also advised to use formula rather than breastfeeding. [7] While pharmaceutical companies and governments continue to funnel millions into vaccine research, there is still no vaccination available to prevent HIV infection. [3] REFERENCES 1. Wiley JM, Sherwood LM, Woolverton CJ. McGraw Hill; 2009 Prescott’s Principles of Microbiology. 1st Ed. Boston: 2. Greene, Warner. AIDS and the Immune System: Scientific American; 1993. 3. Stine GJ. AIDS Update 2008. Boston: McGraw Hill; 2008. 4. What is HIV? [Online] [Accessed March 5, 2010]. Available from: http://www.avert.org/hiv.htm 80 5. HIV: Basic Statistics. CDC. [Online] [Accessed March http://www.cdc.gov/hiv/topics/surveillance/basic.htm#hivest 6. AIDS Orphans. [Online] http://www.avert.org/aidsorphans.htm [Accessed April 15, 6, 2010]. 2010]. Available from: Available from: 7. Schackman BR, Gebo KA, Walensky RP, Losina E, Muccio T, Sax PE, Weinstein MC, Seage GR, Moore RD, Freedberg KA. The lifetime cost of current Human Immunodeficiency Virus care in the United States. Medical Care. 2006; 44: 990 – 997. 8. Preventing Mother to Child Transmission. Avert.org [Online][Accessed March 8, 2010]. Available from: http://www.avert.org/motherchild.htm 81 Anthrax: an acute disease caused by Bacillus anthracis Tracey Coleman General Microbiology, Spring 2010 GENERAL DESCRIPTION OF THE AGENT Anthrax is an acute disease caused by Bacillus anthracis, a spore-forming Gram-positive bacillus. Most forms of the disease are lethal. There are three forms of the disease depending on the route of infection. Forms include inhalation anthrax which causes respiratory infection in humans followed by severe, often fatal, respiratory collapse. [1] Gastrointestinal anthrax produces gastrointestinal difficulty, vomiting of blood, severe diarrhea, acute inflammation of the intestinal tract, and loss of appetite. Lastly, cutaneous anthrax produces boil-like skin lesion that eventually forms an ulcer with a black center known as an eschar. [1] This highly infectious disease typically affects animals. Humans have a high resistance to Bacillus anthracis. Risks of human-to-human transmission arise from animal products imported from endemic regions and from contaminated livestock. There are however, many preventive measures that are taken in these situations. SIGNIFICANCE B. anthracis can form dormant spores that are able to survive harsh conditions over long periods of times, thus destroying the bacterium is extremely important as well as difficult. Anthrax does not spread directly from one infected animal or person to another; it is spread by spores which can be transported by clothing, skin, hide, etc. [2] Anthrax usually infects humans following exposure to infected animals or contaminated animal products. Contact with infected tissues of dead animals such as butchering, preparing contaminated meat, consumption of contaminated undercooked meat, or contact with contaminated hair, wool, or hides. The mortality rate in cutaneous anthrax is 5%-20%, in inhalation anthrax, the mortality rate is 80%-100% and in intestinal anthrax the mortality rate is 25%-75%. [2] Cases of anthrax are extremely rare in the United States; however, anthrax has been used as a biological weapon against the US in 2001 resulting in an outbreak of cutaneous and inhalation anthrax. [3] This incident proved that although decontamination is possible, it is also time-consuming and costly. Decontamination of the Senate office building of anthrax spores cost $27 million. Decontamination of the postal facilities cost $130 million and took 26 months. [3] Decontamination of anthrax on ranches and in the wild is even more problematic. Carcasses can be burned; however, this process takes up to three days, and may not be ideal for smaller areas. Carcasses may also be buried, however, this process requires expensive tools to bury animals deep enough to prevent resurfacing of spores. Carcasses have been soaked in formaldehyde to kill spores; however, this process has environmental contamination issues. GENERAL SYMPTOMS Cutaneous anthrax, which accounts for more than 95% of cases worldwide, results from infection through skin lesions. [1] Intestinal anthrax results from ingestion of spores, and pulmonary anthrax results from inhalation of spores. 82 Cutaneous anthrax most commonly occurs through contamination of a cut or an abrasion. Some countries, however, have biting flies that may transmit the disease. After 2-3 days of the incubation period, a small pimple or papule surrounded by a ring of vesicles appears at the inoculation site. Over the next few days, the central papule ulcerates, dries, and blackens to form an eschar, characteristic to anthrax. [2, 3] The lesion is said to be painless, however, pus and pain can develop if the lesion becomes infected by another organism. The lesion becomes dangerous if on the face or neck because it may swell and block the airway or develop into secondary meningitis. Left untreated, septicemia, blood poisoning due to an invasion of the bloodstream by virulent microorganism, may develop. Approximately 20 percent of untreated cases of cutaneous anthrax progress to fatal septicemia. [2] Gastrointestinal anthrax is similar to cutaneous anthrax but occurs in the intestinal mucosa. B anthracis most likely invades the mucosa through a preexisting lesion. The first symptoms are nausea, loss of appetite, bloody diarrhea, and fever followed by intense stomach pain. [2, 3] In inhalation anthrax, inhaled spores are transported by alveolar macrophages to the lymph nodes. Spores geminate in the lymph nodes and multiply to initiate systemic disease. First symptoms are similar to the common cold including a sore throat, mild fever, and muscle aches. Later symptoms include cough, chest discomfort, and shortness of breath, tiredness, and muscle aches. [2, 3] Gastrointestinal and pulmonary anthrax are both more dangerous than the cutaneous form because they are usually identified too late for treatment to be effective. PATHOGENESIS The pathogenicity of B anthracis depends on two virulence factors; a poly-y-D-glutamic acid polypeptide capsule and a toxin produced in the log phase of growth. [4] The poly-y-D-glutamic acid polypeptide capsule protects it from phagocytosis by the defensive phagocytes of the host. The toxin produced in the log phase of growth consists of three proteins including protective antigen (PA), lethal factor (LF), and edema factor (EF). [4] The protective antigen is a binding protein, which allows the toxin to enter the host cell via endocyte formation. Once inside the endocyte, PA toxin forms a pore, which creates a small passageway in the endosomal membrane that allows the enzymatic components of the toxin to enter the cytoplasm. Edema factor, a calmodulin-dependent adenylate cyclase, combines with PA to form edema toxin. Edema toxin converts adenosine triphosphate to cyclic adenosine monophosphate (cAMP). [4] High intracellular levels of cAMP lead to impaired maintenance of water homeostasis. Edema toxin also inhibits the function of neutrophils and stimulates the production of multiple inflammatory mediators such as neurokinins, prostanoids, and histamines. Rapid release of inflammatory mediators may contribute to the sudden death that can occur with anthrax. Lethal factor, a zinc metalloprotease, combines with PA to form lethal toxin. Lethal toxin stimulates overproduction of cytokines which leads to lysis of macrophages. Lethal toxin has been shown to cause endothelial cell apoptosis and endothelial barrier dysfunction, thus contributing to vascular destruction. [4] Inhalation anthrax Endospores can enter the body through inhalation. Endospores are generally 1µcm x 1.5 µcm in size and can therefore, easily reach the alveoli. Endospores are phagocytosed by macrophages and carried 83 to the lymph nodes or taken up by lung epithelial cells. They begin to germinate inside macrophages and become vegetative cells, leave the macrophages, and multiply in the lymphatic system. [4] B. anthracis enters the bloodstream and precipitates septic shock and toxemia. Septic shock and compression of the lungs are major causes of death. Cutaneous anthrax Endospores are most commonly introduced through the skin via preexisting lesions. Germination will occur at the site of infection and produce localized necrosis with eschar formation and soft-tissue or mucosal edema. The endospores are phagocytosed by macrophages and carried to nearby lymph nodes which cause painful lymphadenopathy (disease of the lymph nodes) and lymphangitis (infection of the lymph vessels). [4] Gastrointestinal anthrax The pathogenesis of gastrointestinal anthrax in not clear because the condition is rare. The site of entry is often the terminal ileum, a section of the small intestine, or the cecum, a portion of the colon. [4] Intestinal lesions occur and result in regional lymphandenopathy. LABORATORY DIAGNOSIS The clinical diagnosis of anthrax is confirmed by culturing B. anthracis, and microscopic techniques including Gram-staining and a direct fluorescent antibody (DFA) test. In inhalation anthrax, if respiratory symptoms are present and sputum is being produced, a specimen can be obtained. [5] Sputum is matter that is expelled from the respiratory tracts including mucus or phlegm. Blood cultures usually do not become positive until the later stages of the disease when treatment is less effective. Cutaneous anthrax can be diagnosed by Gram-staining and culture of vesicular fluid. The best time to collect specimen is during the vesicular stage in which the organism is best demonstrated. To obtain a specimen, sterile swabs should be soaked in vesicular fluid from a previously unopened vesicle. During the eschar stage, sterile swabs should be rotated beneath the edge of the eschar without actually removing the eschar. In gastrointestinal anthrax, stool cultures can be obtained in early stages. In later stages, blood culture can yield the organism, again if drawn before antibiotic treatments. [5] Culture is the definitive test for anthrax. Sputum, swab, and stool specimen should be streaked for isolation on sheep blood agar (SBA) plates. Cultures should be incubated at 35-37°C under ambient conditions. After incubation of SBA plates for 15-24 hours, well-isolated colonies of B. anthracis will be 2-5 mm in diameter. The colonies will appear flat or slightly convex and irregularly round. Edges are slightly undulate and have a ground-glass appearance. Colonies tend to have a firm consistency. If touched with an inoculating loop, the growth will spike up like beaten egg whites. B. anthracis grows rapidly; individual colonies can appear within 12-15 hours. This is characteristic of B. anthracis and can be distinguished from mixed cultures that may contain slower-growing organisms. [5] A Gram stain can be performed on vesicular fluid and blood specimen via usual procedures. B. anthracis will appear as a large, Gram-positive rod, varying from 1-1.5 X 3-5 μm in size forming oval, central to sub-terminal spores on sheep blood agar (SBA). Absence of spores should not rule out B. anthracis because spores may not appear within 24 hours. Vegetative cells seen on Gram stain of blood and 84 smears appear in short chains of 2-4 cells with square ends, characteristic of the bacteria. The presence of large encapsulated gram-positive rods in blood is strongly indicative for B. anthracis identification. [5] A direct fluorescent antibody (DFA) test is a rapid staining technique which tests for the capsules and the cell wall polysaccharides. If both stains are positive, B. anthracis is confirmed. [5] TREATMENT Treatment of anthrax includes antibiotics. People diagnosed with inhalation or gastrointestinal anthrax are initially given IV therapy, which is the first line of defense. Adults are given 400 mg of ciprofloxacin every 12 hours or 100 mg of doxycyclin every 12 hours, in addition to one or more antimicrobials that have in vitro activity including rifampin, penicillin, ampicillin, chloramphenicol, impenem, clindamycin, and clarithromycin. The addition of antimicrobials depends on the course of the anthrax infection. Intravenous therapy is then switched to oral therapy or in combination with each other over the next 60 days. An oral regimen includes 500 mg of ciprofloxacin taken twice daily and 100 mg or Doxycycline taken twice daily. [5] Initiation of therapy with antibiotics during early stages is associated with improved survival. However, patients who progress to later stages have a very high case-fatality rate of 97 percent regardless of therapy. People diagnosed with cutaneous anthrax are prescribed an oral regimen of 500 mg of Ciprofloxacin twice daily or 100 mg of Doxycycline twice daily for 60 days. [6] Treatment does not prevent the development of the skin lesions; however, it will prevent the progression to systemic disease. Cutaneous anthrax cases with signs of systemic disease, extensive edema, or lesions located on the head or neck may require IV treatment and a multidrug approach is recommended. PREVENTION Post-exposure prophylaxis includes 60 days of oral antimicrobial therapy in combination with a threedose series of anthrax vaccine adsorbed (AVA). [7] The vaccine can prevent infection and is given in a series of three injections over 18 months. Currently, the vaccine is not available to the public. It is recommended that only people at high risk of exposure should be given the vaccine including some laboratory workers, people who are in contact with imported animals from other countries, and military personnel. Antimicrobial therapy should be continued for 60 days for people who are exposed to an air space known to be contaminated with B. anthracis, air space known to be the source of inhalation anthrax cases, unvaccinated laboratory workers exposed to confirmed B. anthracis cultures where aerosolization has occurred. [7] Antimicrobial therapy will help prevent the symptoms from developing. If given quickly enough, the spores will not have a chance to germinate and cause infection. However, antibiotics to prevent anthrax are strongly discouraged unless there has been direct exposure to anthrax spores. Misuse of the antibiotics will cause the bacteria to become resistant. 85 Although Anthrax is a highly infectious disease, it typically affects animals. Human usually have a high resistance to B. anthracis. The risk of human-to-human transmission is not high if precautions are taken. [2] Risks arise from animal products imported from endemic regions in which these countries now have strict requirements to limit the risks of importation of products contaminated with anthrax spores. It is impossible to prevent livestock from grazing contaminated vegetation or inhalation when anthrax is present. However, to prevent an outbreak and possible transmission to humans, the infected animal can be treated with penicillin or tetracycline early. When possible, dead, infected animals should be burned where they are found. An alternative is to bury the carcass at a depth of 10 feet and cover the carcass with lime. The premises must also be quarantined. [8] REFERENCES 1. Anthrax fact sheet. World Health Organization (WHO) (online). 2001 (cited 2010 Mar 11). Available from: URL: http://www.who.int/mediacentre/factsheets/fs264/en. 2. Inglesby TV, O’Toole T, Henderson DA, et al., for the Working Group on Civilian Biodefense. Anthrax as a biological weapon, 2002 [cited 2010 Mar 10];287:2236-225. Available from JAMA: http://web.archive.org/web/20051225135530/jama.ama-assn.org/cgi/content/full/287/17/2236 3. Inglesby TV, Henderson DA, Bartlett JG, et al., for the Working Group on Civilian Biodefense. Anthrax as a biological weapon. Medical and public health management. 1999; 281: 1735-1745. [Online] [cited 2010 Mar 10]. Available from JAMA: http://web.archive.org/web/20051222183331/http:/jama.amaassn.org/cgi/content/full/281/18/1735. 4. Anthrax: Current, comprehensive information on pathogenesis. [Online] [cited 2010 Mar 13]. Available from: URL: http://www.cidrap.umn.edu/cidrap/content/bt/anthrax/biofacts/anthraxfactsheet.html#_Epidemiology 5. Basic Laboratory Protocol for the Presumptive Identification of Bacillus anthracis. [online] 2001; [cited 2010 Mar 10]. Available from: URL: http://www.bt.cdc.gov/Agent/Anthrax/Anthracis20010417.pdf 6. Update: investigation of bioterrorism-related anthrax and interim guidelines for exposure management and antimicrobial therapy. [online] 2001; [cited 2010 Mar 10]. Available from: URL: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5042a1.htm. 7. Anthrax Q&A: preventive therapy. U.S. Centers for Disease Control and Prevention. [online] 2005; [cited 2010 Mar 10]. Available from: URL: http://www.bt.cdc.gov/agent/anthrax/faq/preventive.asp. 8. Anthrax: What Livestock Producers Should Know. [online] 2000; [cited 2010 Apr 09]. Available from: URL: http://www.cdfa.ca.gov/ahfss/Animal_Health/pdfs/Anthrax_hh_article.pdf 86 Section 3: Laboratory reports 87 Determination of the growth curve of Escherichia coli in batch culture Ndubisi Chikwem General Microbiology, Spring 2010 ABSTRACT The growth characteristic of Escherichia coli was determined in batch culture, by quantifying the number of colony forming units and turbidity. The results of growth rate by both methods were similar at low turbidity; however, at higher turbidity, the spectrophotometric measurement was less sensitive. It appears that the cultures were in exponential phase throughout the duration of the experiment. This observation was expected because the experiment was only carried out for less than four hours, the duration of the laboratory class. The generation time was calculated to be 22.6 minutes and this is comparable to the generation time of 20 minutes, reported by other researchers. INTRODUCTION Bacteria divide by binary fission, resulting in a rapid increase in cell population. [1] Because of the rapid rate of division, and the single-celled property of bacteria, studies including the determination of pathways are carried out with bacteria. Bacterial growth determination is also important for evaluating industrial cultures and compounds that affect bacteria growth. [2] Consequently, the growth characteristic of bacteria is commonly studied. Every bacterium has its specific grow rate; for example while Escherichia coli divides every twenty minutes, Mycobacterium tuberculosis grows very slowly, dividing every 20 hours. The growth of microorganisms is studied by different techniques including the determination of cell number or the turbidity measurement. While the determination of cell numbers can be tedious and time consuming, it is more accurate in that it measures the number of living organism. On the other hand, turbidity measurement is fast, but not as sensitive. [3] There are four phases of growth for a microorganism in batch culture; lag, exponential, stationary, and death. Lag phase is the beginning phase when the organism is introduced to a new culture so there is no growth; however, the microbe is synthesizing enzymes for the utilization of the nutrients and getting used to the environment. During exponential phase, the organism divides at the maximal rate because all the factors necessary for its growth are provided and nutrient concentration is optimal. Most antibiotics are active against bacteria during the exponential phase since the organisms are highly metabolic at this time. With time, the number of microbes becomes very high; therefore, nutrient is becoming depleted while the level of toxic wastes is getting higher. Consequently, the bacteria enter into the stationary phase in which the number of bacteria dividing is equal to the number of bacteria dying. In the final phase of growth, the phase of death, the organisms dying outnumber those multiplying. Endosporeforming bacteria initiate the process of sporulation at this time. In this experiment, the growth characteristic of Escherichia coli will be determined in batch culture. METHODS E. coli was grown in batch culture by inoculating 50 ml of an overnight broth culture in Brain Heart Infusion into 250 ml of fresh broth that has been equilibrated at 37oC. The culture was placed into a shaking water bath and incubated at 37oC at a speed of 90 RPM. Every 30 minutes, a sample was taken 88 for analysis. A volume of 0.1 ml of the broth culture was diluted in sterile saline to achieve dilutions of 10-2; 10-3 10-4; 10-5 and 10-6. 0.1 ml of each dilution was spread-plated on Brain Heart Infusion Agar in duplicates. The plates were allowed to dry and then incubated at 37 oC for 18 hours, after which plates showing between 50 and 300 colonies were counted. The absorbance of the culture was also determined spectrophotometrically every 30 minutes at 660 nm. A growth curve was prepared using the data from the colony counts and the absorbance measurement. The generation time was calculated using the slope of the curve. RESULTS The results indicate that the generation time of E. coli under the experimental condition is 22.6 minutes. The results also show that the growth curve with absorbance measurement is identical to that of the colony count. However, as the absorbance increased, variance was noticed between absorbance and colony counts. The organism also was in the exponential phase during the experiment and there was no lag or stationary phase. Table 1: Absorbance and CFU/ml of cultures at different times Time (minutes) Absorbance CFU Dilution (660 nm) Counted 0 30 min 60 min 90 min 120 min 150 min 0.08 0.175 0.415 0.707 0.944 1.088 63.5 117 36 63 94 326 10-5 10-5 10-6 10-6 10-6 10-6 Figure 1: Plot of Absorbance against Time 89 Volume Plated CFU/ml 0.1 ml 0.1 ml 0.1 ml 0.1 ml 0.1 ml 0.1 ml 6.35X107 1.17X108 3.6X108 6.3X108 9.4X108 3.26X109 Figure 2: Plot of CFU/ml against time Mean growth rate constant: = 2.65 gen/hour Generation time: DISCUSSION It can be seen from the two graphs (figures 1 & 2) that the growth of E. coli was in the exponential phase throughout this experiment. This is because the starting culture was in BHI broth and the broth in the shaking water bath was already equilibrated at 37oC before the start of the experiment, so the organism did not need a lag phase. The two graphs show near linear increase of growth with absorbance and colony counts. However, it can be seen on the absorbance against time graph that although the bacterial culture was about to enter the stationary phase, it was still in the exponential phase with the CFU/ml against time. This means that the colony count is more sensitive than the absorbance readings as the number of bacteria increased. It would therefore be necessary to dilute the broth when the absorbance was over 0.5 before taking absorbance measurements. When the data was then used to calculate the generation time, it was found to be 22.6 minutes. The known generation time of E. coli is 20 minutes; [1] therefore, the result is similar to that of other researchers. 90 REFERENCES 1. Willey J, Sherwood L & Woolverton C Prescott's Principles of Microbiology. New York: McGraw-Hill Higher Education; 2009. 2. Breidt F, Romick TL and Fleming HP. A rapid method for the determination of bacterial growth kinetics. Journal of Rapid methods and automation in Microbiology. 1994; 3: 59-68. 3. Dominquez MC, de la Rosa M and Borobio MV. Application of a spectrophotometric method for the determination of post-antibiotic effect and comparison with viable counts in agar. Journal of Antimicrobial Chemotherapy. 2001; 47: 391-398 91 Isolation of Klebsiella pneumoniae and Enterobacter aerogenes colonies and examination of morphological characteristics Stephanie Rand General Microbiology, Spring 2010 ABSTRACT A four quadrant streak method was used to isolate Klebsiella pneumoniae colonies on a sterile BHIA plate and characteristics of the morphology were observed and recorded. The 0.7-2.0 mm colonies of Klebsiella pneumoniae were observed to be round, smooth and entire, convex, a creamy/white pigment, and sticky/wet. The 1.0-2.0 mm colonies of Enterobacter aerogenes were observed to be round, entire and smooth, convex, white/light grey, and slightly sticky/wet. Therefore, the colonies were identical in shape, margin, and elevation, and could be differentiated by pigment, texture and size. INTRODUCTION Stock cultures of bacteria often do not contain genetically identical bacteria. Either multiple bacteria exist within the culture, or random mutations may have arisen in a previously isolated bacteria culture. [1] Both the spread plate technique and the streak plating method can be used to isolate bacterial colonies for observation of morphological characteristics. While the spread plate technique allows for fairly accurate enumeration of bacterial colonies, it requires further dilutions of broth and plate cultures. While less accurate, streak plating is a less complicated process, and is more conducive to a student lab. [2] In this lab, the streak plate method was used to isolate colonies from broth cultures of Klebsiella pneumoniae and Enterobacter aerogenes. This allowed for the study of morphological characteristics including shape, margin, elevation, texture, pigment production and size. [3] MATERIALS AND METHODS Part I: Inoculation of Agar Plates Using the Quadrant Streak Method A loop was sterilized with a Bunsen burner, and then used to transfer samples of Klebsiella pneumoniae and Enterobacter aerogenes cultures to sterile BHIA (Brain-Heart Infusion Agar) plates. Each sample was gently (so as not to cut the agar) dragged in a zigzag pattern across ¼ of the plate’s surface. During inoculation, the lid of the agar plate was used as a shield to prevent airborne contamination. After inoculation, the lid was replaced. The loop was sterilized again using the Bunsen burner. The loop was allowed to cool for 10 seconds. Another streak was created on the agar plate, starting in the well, and moving over to include another ¼ of the agar plate. The loop was sterilized and cooled again, and used to create a third streak beginning in the second streak. The loop was sterilized and cooled again, and used to create a fourth streak beginning in the third streak. The fourth streak was extended into the middle of the plate, consuming the rest of the agar plate, while not entering the first, second or third streaks. The loop was sterilized again before being put away. The plate was labeled and incubated at 37°C for 18 hours in an inverted position. Part II: Determining Colony Morphology of Bacterium by Observation of Streaked Agar Plates The BHIA plates, previously inoculated with cultures of Klebsiella pneumoniae and Enterobacter aerogenes were examined. Bacterial colony growth was observed with both the naked eye and with the assistance of a magnifying glass. A metric ruler was used to measure the size of the colonies. The characteristics of the respective colonies were observed and recorded in Table 1.1 (see below in 92 Results). Colonies were examined with respect to size, shape, elevation, margin, texture and pigmentation. The differences between Klebsiella pneumoniae and Enterobacter aerogenes were noted. RESULTS COLONY FEATURES Table 1 Observations of Agar Plate Streaked With K. pneumoniae and E. aerogenes WHOLE COLONY MARGIN ELEVATION PIGMENT TEXTURE SIZE (mm) OBSERVATIONS Klebsiella pneumoniae Round Smooth, entire convex Cream/white Sticky, appears moist/wet and opaque 0.7-2.0 mm Enterobacter aerogenes Round Smooth, entire convex White/light grey Slightly sticky/wet 1.0-2.0 mm The results show that colonies of both bacteria were different in size, with those of Klebsiella pneumoniae being slightly smaller. The organisms also differed with respect to texture and pigmentation. The shape and elevation of both organisms were however, identical. DISCUSSION The streak plate technique resulted in the isolation of discrete colonies of Klebsiella pneumoniae and Enterobacter aerogenes for observation of morphological characteristics. While identical in shape, margin, and elevation, the two colonies differed in pigmentation, texture and size. While differentiation would be difficult between Klebsiella pneumoniae and Enterobacter aerogenes, texture and pigment could be used for discriminating between the two. Size should not be used to differentiate the two, as it only differed slightly (± 0.3 mm). The streak plate technique would enable researchers to obtain discrete colonies from a mixed culture. REFERENCES 1. Center for Polymer Studies at Boston University. Bacterial Growth: Streaking and Inoculating Plates. [Online] [Cited 2010 Jan 21]. Available from: http://polymer.bu.edu/ogaf/html/chp51exp2.htm 2. Experiment 7: Enumeration of Bacteria. [Online] [Cited 2010 Jan 21]. Available from: http://as.clayton.edu/furlong/BIOL3250/lab/objectives_instructions/Labs/Bacterial%20Enu:meration08. pdf 3. Microbiology: Laboratory Theory and Application; Fundamental Skills for the Microbiology Laboratory and Microbial Growth. Lab Handout 93 Simple and Gram staining of Klebsiella pneumoniae and Enterobacter aerogenes Brian Cooper General Microbiology, Spring 2010 ABSTRACT Cultures of Enterobacter aerogenes and Klebsiella pneumoniae, were smeared onto clean glass slides, heat fixed, and stained by Gram and simple stain. Microscopic examination of the stained smears revealed that Enterobacter aerogenes appeared pink after the gram-stain, identifying it as a Gramnegative organism while Klebsiella pneumoniae also appeared pink, thus showing that it is also a Gramnegative organism. Simple staining revealed that both organisms are rods, spread out, and stained blue, the color of the primary stain. Enterobacter aerogenes rods were found to be shorter than those of Klebsiella pneumoniae. INTRODUCTION When observing a specimen by microscopy, it is essential for the specimen to contrast with the background of the microscope field. Without stains to provide this contrast, observing cells can be very difficult. Often times, stains are used to observe cell morphology, size and arrangement. The morphologies of cells can be spheres (cocci, singular coccus), rods (bacilli, singular bacillus), or spirals (spirilla, singular spirillum). Slightly curved rods are vibrios, short rods are coccobacilli, and flexible spirals are spirochetes. The arrangement of cells is related to cell division which could be singles, pairs, cubes, tetrads or chains. Two types of stains are commonly used in a Microbiology laboratory. One such stain is a simple stain. Simple stains contain a solvent (usually water or ethanol) as well as a colored molecule, known as the chromogen. The chromophore gives the chromogen its color. The auxochrome is the charged portion of a chromogen and allows it to act as a dye through ionic or covalent bonds between the chromogen and the cell. [1] In simple stains, the auxochrome becomes positively charged by picking up a hydrogen ion or losing a hydroxide ion. Therefore, positive simple stains are attracted to the negative charges on the surface of most bacterial cells. [1] The other type of stain, differential stains allows for observation of differences between organisms or the differences between parts of the same organism. A type of differential stain, Gram stain, has a decolorization step that occurs between two basic stains. The primary stain is crystal violet while iodine is added as a mordant to enhance crystal violet staining by forming a crystal violet-iodine complex. Decolorization occurs after the addition of iodine and is normally done with alcohol, acetone, or ethanol. Gram-negative cells are decolorized by the solution while Gram-positive cells are not. A counterstain of safranin is applied to color the Gram-negative cells. Upon completion, Gram-positive cells appear purple while Gram-negative cells appear reddish-pink. Since Gram-negative cell walls have a higher number of lipids and thinner peptidoglycan layer than Gram-positive cell walls, the alcohol/acetone/ethanol extracts the lipid, making the cell more porous and therefore causing it to not retain the crystal violet-iodine complex, decolorizing it. Gram-positive walls are less susceptible to 94 decolorization due to its thicker peptidoglycan layer and cross linking. [1] Upon completing these stains, it should be possible to determine the characteristics of both Enterobacter aerogenes and Klebsiella pneumoniae. MATERIALS AND METHODS Bacterial smears The cultures of Enterobacter aerogenes and Klebsiella pneumoniae were obtained from the instructor. After lighting a Bunsen burner, a metal loop was sterilized in the flame. This loop was then dipped into a beaker of water and a small drop of water from the loop was placed on a clean glass slide. After sterilizing the loop once again, it was delicately used to obtain bacterial growth from an isolated culture. The loop was then placed on one of the drops of water. The bacteria were mixed in and the water was spread out to produce smears. This process was carried out for both organisms and for both stains. The smears on the slides were allowed to dry in air. Upon drying, a slide holder was used to pass the smear through the upper part of a flame two or three times. This heat-fixing kills the bacteria, makes them adhere to the slide, and coagulates cytoplasmic proteins to make them more visible. [1]The slide was allowed to cool before the performance of the staining. Simple staining The slides were placed on a slide rack positioned over a staining tray. The smears were covered with the crystal violet stain. After letting the stain sit for 1 minute, the stain was rinsed off with water. The slides were gently blotted dry using bibulous paper. Gram Staining The slides with the heat-fixed smears were placed on a slide rack positioned over a staining tray. The smears were covered with the crystal violet stain. After letting the stain sit for 1 minute, the stain was rinsed off with distilled water. Then the smears were covered with Lugol’s Iodine and left for 1 minute, after which, the iodine was rinsed off with water in the same way as was done for the crystal violet. The smears were then decolorized with alcohol until the run-off was clear. It was essential that this was only done for a few seconds to ensure that the cells weren’t over-decolorized which could cause inaccurate results. The slides were thoroughly rinsed with water once again. Next, counterstaining was done with Safranin stain and allowed to sit for one minute. After rinsing with water, the slides were gently blotted dry using bibulous paper. Microscopic examination of stained slides Using an oil immersion lens and a bright field microscope, the stained slides were observed and the cell morphology, arrangement, and size were recorded. RESULTS Both organisms appeared to have been stained by both the simple and gram stains. The cell morphology, size, and arrangement of the Enterobacter aerogenes and Klebsiella pneumoniae are below in Table 1. The Gram reactions of both organisms are also shown in Table 2. The results show that both 95 organisms are Gram negative bacilli that appear singly. The cells of Enterobacter aerogenes are shorter than those of Klebsiella pneumoniae and therefore tend to appear as coccobacilli. Table 1: Cell Characteristics for Simple Stain. Organism Size Morphology (Shape) Arrangement Enterobacter aerogenes Short (slightly smaller the Kp) Rod (possibly coccibacilli) Spread out Klebsiella pneumoniae Long Rod Spread, un attached upon division Table 2: Cell color after Gram-Stain. Organism Color Gram type Enterobacter aerogenes Pink Gramnegative Klebsiella pneumoniae Pink Gramnegative DISCUSSION Enterobacter aerogenes appeared pink after the gram-stain, therefore, it is a Gram-negative organism. Klebsiella pneumoniae is also a Gram-negative organism since it appeared pink. Both organisms were found to be rod shaped and spread out. Enterobacter aerogenes rods were found to be shorter than those of Klebsiella pneumoniae. In future, it would be necessary to include Gram positive and Gram negative organisms of different shapes in this exercise. This would enable students to have more experience with Gram positive organisms that have a tendency to over-decolorize. LITERATURE CITED 1. Bacterial Structure and Simple Stains. In: Microbiology: Theory and Application; Leboffe MJ and Pierce BE eds. Brief ed. Colorado; Morton Publishers. 2008; p. 145-163. 96 Evaluation of Pathogen Sensitivity to Chemotherapeutic Agents Jolie Wax General Microbiology, Spring 2010 ABSTRACT Cultures of Staphylococcus aureus, Bacillus cereus, Escherichia coli, Klebsiella pneumoniae, Enterobacter aerogenes and Pseudomonas aeruginosa were tested for antibiotic susceptibility using the disc diffusion method. After incubation for 24 hours, the zones of inhibition of Ampicillin, Chloramphenicol, Erythromycin, Penicillin, Streptomycin and Tetracycline were measured and related to the table provided by The National Committee for Clinical Laboratory Standards. P. aeruginosa showed the greatest resistance to antibiotics while Staphylococcus aureus and Bacillus cereus were susceptible to most. From this we can conclude that gram staining qualities of bacteria may relate to their antibiotic susceptibility. INTRODUCTION Chemotherapeutic agents are used widely by physicians to treat infectious diseases. Antibiotics are chemotherapeutic agents naturally derived from fungus and bacteria that produce them in order to inhibit the growth of other microbes. Among some of the first discovered were Penicillin and Streptomycin. Penicillin, produced by a mold, was discovered in 1928 by Alexander Fleming, and Streptomycin was isolated by Selman Waksman in 1940 from a soil bacterium. [1] Microbial resistance to antibiotics is an increasing health concern. It is the result of antibiotic additives to feed farm animals, unnecessary prescriptions, not finishing a prescription, and other causes. [2] Bacteria have the ability to mutate rapidly and new pathogenic strains are being discovered. There is always a need for new antibiotics to be discovered and tested. In the laboratory, paper discs absorbed with antibiotics may be used to determine the sensitivity of a particular pathogen that has been isolated. This is important when determining dose concentrations for treatment. The paper discs are inoculated with the chemosynthetic agent. When the discs are placed on a Petri dish inoculated with the microbial pathogen, the chemotherapeutic agent will diffuse and a zone of inhibition can be formed. The size of the zone can be affected by several factors such as salt concentration, depth of the agar and the number of microorganisms present. In efforts to standardize bacterial inhibition measurements by antimicrobial disks, a uniform technique is used in laboratories. The disc diffusion method uses Mueller-Hinton agar which allows the chemotherapeutic agent to diffuse freely. [1] In this laboratory, the disc diffusion method will be used to observe the diameter of the zones of inhibition of six chemotherapeutic agents on Staphylococcus aureus, Bacillus cereus, Escherichia coli, Klebsiella pneumoniae, Enterobacter aerogenes and Pseudomonas aeruginosa. METHODS AND MATERIALS The antimicrobial sensitivity test began with six Mueller-Hinton agar plates. Cultures of bacteria were grown in Brain Heart Infusion broth and incubated for 18 hours at 37 oC. Broth cultures were standardized to 0.5 McFarland before being used for the susceptibility tests. Each plate was streaked using a sterile cotton swab to cover the plate evenly with one of the following bacteria: S. aureus, B. 97 cereus, E. coli, K. pneumoniae, E. aerogenes and P. aeruginosa. While sterilizing forceps between steps, six different antimicrobial discs were placed and lightly pressed into the medium of each plate. The discs were evenly placed, and not too close to the edge. Each plate received one of the following discs; Streptomycin, Erythromycin, Penicillin, Tetracycline, Ampicillin and Chloramphenicol. Plates were inverted and incubated at 35 degrees Celsius for 24 hours. The observed diameter of the zone of inhibition was measured and recorded. RESULTS Table 1 shows the diameter of the zones of inhibition formed by each antibiotic on the six bacteria used in this experiment. The zone measurements are accompanied by either an (r) for resistance, an (i) for intermediate or an (s) for susceptible. The R, I and S values are standardized by The National Committee for Clinical Laboratory Standards. [1] The results show that only Staphylococcus aureus was sensitive to Penicillin, and that Pseudomonas aeruginosa was the most resistant of the test bacteria. Chloramphenicol appeared to be the most effective of all the antibiotics since it killed all bacteria except Pseudomonas aeruginosa. Table 1: Inhibition Zones for Test Cultures; Diameter of Zones of Inhibition (mm) S. aureus B. cereus E. coli K. E. aerogenes P. aeruginosa pneumoniae AM 30 (s) 0 (r) 20 (s) 0 (r) 0 (r) 0 (r) C 25 (s) 30 (s) 33 (s) 30 (s) 23 (s) 0 (r) E 0 (r) 30 (s) 14 (i) 10 (r) 0 (r) 0 (r) P 40 (s) 0 (r) 10 (r) 0 (r) 0 (r) 0 (r) S 25 (s) 23 (s) 10 (r) 25 (s) 14 (i) 15 (i) TE 25 (s) 17 (i) 20 (s) 20 (s) 18 (i) 8 (r) AM (Ampicillin); C (Chloramphenicol); E (Erythromycin); P (Penicillin); S (Streptomycin); TE (Tetracycline) DISCUSSION The experiment showed a lot of diversity in the susceptibility of microorganisms to different antibiotics. In general, the gram positive bacteria were less resistant. All organisms except P. aeruginosa were susceptible to Chloramphenicol. All gram-negative organisms showed resistance to Penicillin. This is expected since Penicillin inhibits the transpeptidation step in cell wall formation. Gram-negative organisms do not form cross walls and therefore not involved in transpeptidation. The resistance of 98 Pseudomonas aeruginosa to the antibiotics used in this experiment is not surprising because it is one of the most difficulty organisms to kill. In future, other antibiotics like gentamicin should be included to determine its effectiveness against Pseudomonas aeruginosa. All of the bacteria used in this experiment are part of our natural bacterial flora, and therefore, have the potential to become a health risk. If a particular organism is valued as susceptible to the dosage of the antimicrobial disc it may be an appropriate treatment for the disease caused by that particular pathogen. It is important to keep an ongoing national record in order to know the level of antibiotic resistance by new strains of common pathogens. It is also important to perform antimicrobial susceptibility of isolates before treatment in order to avoid the possibility of over-exposure of microbes to antibiotics that could eventually lead to resistance. REFERENCES 1. Mayo, William J., Chemical Methods of Control: Antimicrobial Drugs. Exercise 25. Laboratory handout. 2010. 2. Todar, Kenneth. The Microbial World: Bacterial resistance to antibiotics. 2009. [Online] [Accessed march 19, 2010]. Available at: http://textbookofbacteriology.net/themicrobialworld/bactresanti.html 99 Determination of Water Quality by Presumptive, Confirmed, and Completed Tests Wydia Davis General Microbiology, Spring 2010 ABSTRACT Two samples of water were tested by the presumptive, confirmed and completed tests. Sample A had no fecal coliforms and was therefore suitable for drinking. Sample B on the other hand had significant numbers of coliform bacteria, and therefore, not suitable for drinking. The result of this experiment confirms that sources of drinking water should be tested periodically for their suitability for domestic use. INTRODUCTION Water is commonly used in our homes, businesses and environment. It is therefore extremely important to determine whether it is safe to drink and /or use. By the late nineteenth century, microbes had been linked to diseases such as typhoid, dysentery and cholera, and fecal contamination had become the basis for the development of public water treatment. [1] Water quality can be determined by the kinds of microorganisms found in the water samples. It would be very difficult to test water for the organisms that are known to cause disease including Vibrio cholerae, Salmonella typhi or Shigella dysenteriae because they may be in such small numbers, fastidious and time consuming to culture individually. [2] Instead, Escherichia coli is used as an indicator of water contamination. E. coli may be defined as a coliform, gram-negative, non-endospore forming, facultative anaerobic rod that ferments lactose and produces gas within 48 hours when incubated at 35 oC. E. coli is normally found in the intestines of warm-blooded animals and not in soil or water. It is not fastidious and can survive for several days in the water, making it a good indicator of recent contamination. [2] Finding E. coli in water is therefore indicative that the water has recently been contaminated with fecal materials. Currently the EPA tests public water supplies regularly for 90 different contaminants, including E. coli, Salmonella, Cryptosporidium, lead and disinfection byproducts. [3] In this laboratory three tests are performed to determine the contamination of water from two different water sources. The Presumptive Test identifies lactose fermenting gas producing bacteria. The Confirmed Test selects for gram negative bacteria. Finally the IMViC (indole, Methyl Red, Voges-Proskauer & Citrate) tests differentiate members of Enterobacteriaceae. These tests identify strains of E. coli based on; reduction of tryptophan, mixed acid fermentation, and use of sodium citrate. MATERIALS AND METHODS Presumptive Test Two samples of water (A and B) were provided to the students. Each water sample was shaken 25 times to mix. 10 ml of each water sample was aseptically pipetted into each of three tubes containing 10 ml of double strength lactose broth and an inverted Durham’s tube. 1 ml of each water sample was pipetted 100 into each of three tubes containing 10 ml of single strength lactose broth and an inverted Durham’s tube. Finally, 0.1 ml of each water sample was pipette into each of three tubes containing 10 ml of single strength lactose broth and an inverted Durham’ tube. 1.0 ml of each water sample was placed into each of three sterile Petri dishes. Molten Yeast extract agar cooled to 45oC was carefully poured over the sample of water. The plates were swirled ten times to the left, followed by ten times to the right and finally ten times up and down. The plates were left on the table to solidify. The inoculated tubes and plates were appropriately labeled, and then incubated at 35 oC for 48 hours. After incubation, the number of colonies growing on each plate was counted and recorded. The number of tubes indicating lactose fermentation (yellow), with gas (displacement in Durham’s tube) was also counted. Most Probable Number (MPN) was determined by referring to a Probability table. Confirmed Tests Plates of MacConkey agar were streaked with cultures from each tube showing lactose fermentation with gas production. Plates where incubated at 35 oC for 24 hours and observed for lactose fermenting colonies (red/pink colonies). Suspected colonies were Gram-stained for the presence of Gram-negative bacilli. Completed Tests Three colonies from each plate with suspected coliforms (pink colonies on MacConkey agar and Gram negative bacilli) were tested for Indole, Methly-red, Voges-Proskauer and Citrate utilization tests (IMViC). Each suspected colony was cultured into tubes of peptone water, MR-VP broth and Simmon’s citrate slant. The cultures were incubated at 35oC for 24 hours before examination. Indole production was determined by the addition of 5 drops of Kovac’s reagent to a peptone water culture of the isolate. A red color indicates a positive result. The tube culture of MR-PV broth was divided into two. 0.5 ml of methyl red was added to one of the tubes. A red color indicates a positive result. In order to test for the presence of acetoin and 2,3 butanediol (Voges-Proskauer test), 0.5 ml of VP reagent was added to the tube. A red color indicates a positive result. To test for the utilization of citrate as the sole source of carbon, the inoculated Simmon’s citrate slant was examined for change of color from green (negative) to blue (positive). RESULTS There was no acid and gas produced in tubes containing water sample A. Therefore, no further tests were required to confirm the presence of coliforms in this water sample. All tubes containing water sample B showed acid and gas production. The most probable number (MPN) of coliforms in sample B was determined from the Probability table as 1,100 coliforms per 100 ml of 101 water. All the tubes were therefore subjected to the confirmed and completed tests. The number of colonies growing from the pour plate of sample B was counted and averaged 76 colonies per ml. The plates of MacConkey agar showed typical fecal coliform colonies. Colonies appeared as convex and pinkish, indicating that they are lactose fermenting, gram negative bacteria. Gram staining of the colonies confirmed that the colonies are gram-negative bacilli. The addition of Kovac’s reagent to the peptone water cultures turned the sample red indicating the production of indole. The addition of methyl red also produced a red color. However, the VP test and citrate utilization tests were negative. As a result of these results, we concluded that water sample B contained fecal coliforms. DISCUSSION It is clear from the results that sample B was highly contaminated because all the lactose broth tubes showed the presence of lactose fermenting and gas producing bacteria. The confirmed and completed tests also proved beyond any reasonable doubt that the sample of water was heavily contaminated and therefore unsuitable for domestic use. We could not ascertain the origin of the water sample since they were given to us by the instructor. In future, it would be necessary to include volumes of 0.01 in the test protocol in case the water sample is heavily contaminated as was the case with sample B. REFERENCES 1. Excel Water Technologies Inc. 2007. The History of Drinking Water Treatment. [Online] [Accessed March 19, 2010]. Available on the web: http://www.excelwater.com/eng/b2c/about_7.php 2. Bacteriological Examination of Water: Qualitative Tests. 2010. Laboratory handout for General Microbiology. 3. Centers for Disease Control and Prevention. [Online] [Accessed march 19, 3010]. Available on the web: http://www.cdc.gov/healthywater/drinking/public/water_quality.html 102 Staining of Mycobacterium smegmatis for acid fastness and Bacillus cereus for endospores Laura Brown General Microbiology, Spring 2010 ABSTRACT A smear of M. smegmatis was stained by the Ziehl-Neelsen technique to determine if the M. smegmatis was an acid fast bacterium. Another smear of B. cereus was stained by the Shaeffer-Fulton technique to determine if the bacterium was an endospore producer. Microscopic examination of the acid fast stain revealed clusters of red-stained, slender bacilli, thereby confirming that M. smegmatis is acid fast. The smear containing B. cereus also showed red-stained bacilli in chains and pairs; however, some of the cells contained oval green spores that were sub-terminal in location. We therefore conclude that B. cereus is an endospore former. INTRODUCTION Staining of bacteria can be carried out by several techniques including the Ziehl Neelsen’s acid fast stain and the Shaeffer-Fulton’s method for endospores. These techniques are very useful for detecting structures that are difficult to stain by conventional staining techniques. Some bacteria have waxy cell walls materials made of mycolic acid. This lipid material makes the organisms resistant to penetration by ordinary dyes. Through heating and the use of carbol fuchsin, the dye can penetrate the cell wall and stain the bacteria. Once stained, these bacteria resist decolorization even with acid alcohol. Acid fast staining is important in the diagnosis of M. leprae (the cause of leprosy) and M. tuberculosis (the cause of tuberculosis). M. leprae cannot be grown on artificial culture media, and it takes between six to eight weeks for colonies of M. tuberculosis to develop on culture media. Therefore, demonstrating acid fast organisms in appropriate clinical samples is diagnostic for these two pathogens. Spores are produced mostly by gram positive bacilli including those of the genera, Clostridium and Bacillus, which makes their identification by staining difficult. This spore-coat is very thick, resistant to many toxic molecules, and contains enzymes that detect favorable conditions for growth. Endospores ensure the survival of bacterium through periods of environmental stress, because they are resistant to UV radiation, temperature change, starvation, etc. Endospores are resistant to staining by ordinary dyes; however, when these spores are subjected to harsh conditions such as heating and the use of malachite green, the dye can penetrate and stain the spores. The spores remain stained even after decolorization with water. This staining technique is important in the diagnosis of Clostridium tetani (tetanus) and B. anthracis (anthrax). The position of the spores within the cell and the spore shape are genetically determined and therefore important in the diagnosis of endospore formers. In this laboratory, the Ziehl Neelsen’s stain will be used to determine if M. smegmatis is acid fast. The Shaeffer-Fulton’s method will also be used to determine if the culture of B. cereus produced endospores. 103 METHODS AND MATERIALS Colonies of two bacteria, M. smegmatis and B. cereus were provided along with glass slides. A wire loop was sterilized and cooled, and then used to place a drop of water on the slide. The wire loop was flamesterilized, cooled, and used to scrape a colony of the organism. The loop was used to produce smears of the bacteria in the drop of water on the slide. After preparing the smears on the glass slide, it was allowed time to dry and then heat fixed. The slides were placed on a staining rack and stained. The slide with the M. smegmatis smear was covered with hot carbol fuchsin. Heat was applied with a cotton swab under the slide until vapors rose, and the stain was left for 10 minutes. The smear was allowed to cool, then washed with water and decolorized with acid alcohol. The smear was then counterstained with methylene blue for one minute, rinsed with water and allowed to dry. The slide containing the smear of B. cereus was stained with Malachite green and heated until vapor rose. The stain remained on the slide for ten minutes, after which it was thoroughly rinsed with water and counterstained with Safranin for one minute. The slide was then washed with water and allowed time to dry. The slides were then observed with the oil immersion lens. RESULTS Following the procedure, the slides were examined for the stained organisms using the oil immersion lens. The slide containing B. cereus showed large red bacilli, some in pairs and others in chains. Green stained, oval spores were found in the cells, while some spores were free on the slide. The spores inside the cells were sub-terminally located. This indicates that B. cereus is a spore former. The slide containing M. smegmatis showed red stained bacilli in clusters. No other forms were seen and this signifies that M. smegmatis is an acid fast bacterium. DISCUSSION The acid fast (Ziehl Neelsen) and endospore stain allowed us to distinguish which type of bacterium we were dealing with in a short time period. We were able to see vegetative cells, endsopores within cells and free spores. The same was true for the M. smegmatis staining where the organisms were seen in clusters typical of Mycobacterium. It would be recommended that negative controls of non-spore formers and non-acid fast organisms should be included in future laboratories. Given the diagnostic nature of the acid fast technique, it is a very useful staining procedure for any microbiology student. REFERENCES 1. Acid fast stain. [Online] [Cited 2010 March http://ftp.ccccd.edu/dcain/CCCCD%20Micro/acid_fast.htm 3]. Available from : URL: 2. Acid fast stain. [Online] [Cited 2010 March 3]. http://ftp.ccccd.edu/dcain/CCCCD%20Micro/endospore.htm Available from : URL: 104