Structural Characterisation of Cationically Modified Trimyristin
advertisement

Rheinisch-Westfälische Technische Hochschule Aachen
Fakultät für Mathematik, Informatik und Naturwissenschaften
Structural Characterisation of
Cationically Modified Trimyristin Nanoparticles
and their Complex Formation with DNA
Strukturelle Charakterisierung von kationisch modifizierten
Trimyristin-Nanopartikeln und ihre Komplexbildung mit DNA
Masterarbeit
zur Erlangung des Grades eines Masters of Science
der Fakultät für Mathematik, Informatik und Naturwissenschaften
an der RWTH Aachen
vorgelegt von
Charlotte Knittel
aus Aachen
11. Januar 2013
Contents
1 Introduction
1.1 Background . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2 Objective . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2 Theory
2.1 Differential scanning calorimetry (DSC)
2.2 Small Angle X-Ray-Scattering (SAXS) .
2.2.1 General considerations . . . . . .
2.2.2 Scattering of a dispersion of lipid
2.3 Photon correlation spectroscopy (PCS) .
2.4 ζ-Potential . . . . . . . . . . . . . . . . . .
. . . . . . . .
. . . . . . . .
. . . . . . . .
nanocrystals
. . . . . . . .
. . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
3 Experimental section
3.1 Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.2 Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.2.1 Photon correlation spectroscopy measurements (PCS) . . . .
3.2.2 Small and wide angle X-ray scattering measurements (SAXS
WAXS ) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.2.3 DSC measurements . . . . . . . . . . . . . . . . . . . . . . . . .
3.2.4 ζ-Potential measurements . . . . . . . . . . . . . . . . . . . . .
3.3 Preparation of the nanodispersions . . . . . . . . . . . . . . . . . . . .
3.3.1 Ultrasonication . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.3.2 High pressure melt homogenisation . . . . . . . . . . . . . . . .
3.3.3 Preparation of DNA-trimyristin nanoparticle-complexes . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
. . .
. . .
. . .
and
. . .
. . .
. . .
. . .
. . .
. . .
. . .
4 Results and discussion
4.1 Trimyristin nanodispersions prepared by ultrasonication . . . . . . . . . .
4.1.1 Preparation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.1.2 Nanodispersions containing Poloxamer . . . . . . . . . . . . . . . .
I
1
1
5
8
8
11
12
14
15
18
20
20
21
21
21
23
23
23
23
25
26
27
27
27
28
Contents
4.2
4.3
4.1.3 Poloxamerfree Nanodispersions . . . . . . . . . . . . . . . . . . . . .
Trimyristin nanodispersions prepared by high pressure melt homogenisation (HPMH) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2.1 Preparation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.2.2 SAXS and WAXS measurements . . . . . . . . . . . . . . . . . . . .
4.2.3 DSC measurements . . . . . . . . . . . . . . . . . . . . . . . . . . . .
DNA complexes of cationic modified triglyceride nanosuspensions . . . .
4.3.1 Preparation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.3.2 SAXS measurements . . . . . . . . . . . . . . . . . . . . . . . . . . .
4.3.3 DSC measurements . . . . . . . . . . . . . . . . . . . . . . . . . . . .
30
32
32
34
39
42
43
43
47
5 Conclusion and future prospects
53
Bibliography
55
Appendix
60
II
Abstract
Triglyceride nanoparticles have been proposed as a new type of drug carrier system
for the administration of poorly water soluble drugs. Promising results obtained with
tripalmitin dispersions and their DNA complexes encouraged further exploration. So
far, stable dispersions of cationically modified trimyristin nanoparticles have only been
obtained in the presence of phospholipids and an additional co-stabilizer. In this thesis
it was attempted to prepare stable dispersions without additional co-stabilizers and to
study their DNA complex formation, which has not been reported yet.
A series of experiments has been conducted to compare two different preparation methods for the particles (high pressure melt homogenisation and ultrasonication). Stable
dispersions with nanoparticles in the size range of 100 nm and 220 nm could be prepared
by both methods. The dispersions were characterised by PCS, SAXS, WAXS and DSC.
Characterisation of the dispersions suggests a particle self-assembly into stacks and multiple discrete melting events, which can be accounted to a particle size effect known from
literature.
The attempt to produce DNA-trimyristin nanoparticle-complexes led to agglomeration
and no reproducible and stable complexes could be obtained. SAXS pattern show unknown features indicating that no self-assembly into stacks took place as for the native
dispersions and a possible interpretation would be the formation of a different structure.
DSC curves show the transformation of multiple discrete melting events into a single
bulk like melting event with increasing DNA content. Previous work suggests that an
additional stabilizer is required to allow a stack formation and stable complexes.
Overall, these results suggest an influence of the co-stabilizer on physical parameters like
the particle size and the crystalline composition and thus on the complex formation. A
higher amount of co-stabiliser increases the particle size and displays a different complex
formation. The influence of the chain length of the co-stabilizer is observed as the particle
size decreases with decreasing length and ratio of the crystalline phases changes as the
amount of the first observed phase decreases and the β-phase respectively changes.
III
Contents
DSC and PCS are indirect methods and need complementary methods in terms of information on the structure of the sample. SAXS gives direct structural information,
but complimentary methods help with the interpretation. Therefore Cryo-transmission
electron microscopy represents a suitable complimentary method as lipid nanoparticles
can be studied and further investigations would allow deeper insights on the structure
of the pure dispersions and their DNA complexes and also confirm the suggestions made
throughout this thesis.
IV
Die vorliegende Masterarbeit wurde im Zeitraum von September 2012 bis Februar 2013
im Arbeitskreis von Professor Tobias Unruh am Lehrstuhl für Kristallographie und
Strukturphysik der Friedrich-Alexander Universität Erlangen angefertigt. Hiermit erkläre
ich, Charlotte Knittel, an Eides statt, dass ich die Arbeit selbstständig verfasst und keine
anderen als die angegebenen Quellen und Hilfsmittel verwendet habe.
Erlangen, 2013
1 Introduction
1.1 Background
The search for suitable delivery systems for the administration of poorly water soluble
drugs into the body has been an important challenge in pharmaceutical research. Colloidal triglyceride emulsions which have already been used as parenteral nutrition for
more then 50 years [1] are a promising application as a drug carrier system. [2,3] The applicability of emulsions is limited due to their uncontrollable release and for this reason
the interest shifted from emulsions to suspensions and specifically to suspensions of lipid
nanoparticles. [4] The incorporation of a drug into a solid core is expected to bind the
drug stronger and to provide a better protection against degradation. A solid core also
offers a higher potential for sustained or controlled drug release and is expected to possess a better physical and chemical stability as compared to liquid or liquid crystalline
carriers. Using lipids as carrier material has the advantage that they are well tolerated
and biodegradable. [2,3,5,6]
Lipid nanoparticles
Dispersions of lipid nanoparticles consist of particles with a solid lipid core (for example,
glycerides, fatty acids, waxes), which is stabilized by a shell of an emulsifier (for example,
phospholipids, polyoxyethylene ether, quaternary ammonium salts) and a dispersion
medium. The most popular preparation methods are either based on the high energy
dispersion of the lipid phase (high pressure melt homogenisation or ultrasonication)
or the precipitation from a homogeneous system. The particle sizes range typically
between 50 and 400 nm. [6] This study is focused on triglyceride nanoparticles stabilized
by phospholipide and another co-emulsifier.
Crystallization behaviour and Polymorphic transitions
Nano-dispersions obtained from molten lipids generally require cooling below a critical
temperature to ensure crystallization. [3,7] For triglycerides, a pronounced supercooling
1
CHAPTER 1. INTRODUCTION
Figure 1.1: Trimyristin in its triclinic crystal structure. [13]
in the dispersed state is observed as the dispersed materials exhibit crystallization temperatures which are much lower than the bulk crystallization temperature of the same
material. Supercooling is much more pronounced in the colloidal state due to the absence
of crystallization promoting impurities within the majority of the dispersed droplets (homogeneous nucleation). [6,8]
Solids of the same composition that can exist in more than one form are referred to as
polymorphic. [9] Triglycerides are polymorphic as they exist in different crystalline phases
and they usually undergo complex processes of monotropic polymorphic transitions after
crystallization. It has been observed for bulk triglycerides that the melt crystallizes in the
metastable α-modification which then transforms eventually via the β’-modification into
the stable β-polymorph of triclinic crystal structure [10] (Fig. 1.1). Each transformation
is a complex molecular reorganization process within the crystal lattice. Triglyceride
nanodispersions are highly dynamic systems as ageing or storage processes continue to
influence the polymorphic transitions after crystallization and the rate depends on the
sample composition. [3,6]
The incorporation of drug substances into the nanoparticles can influence the crystallization behaviour and subsequently the polymorphic transitions. [8,11] One example is the
incorporation of the drug ubidecarenone which may lower the crystallization temperature and accelerates the polymorphic transitions. [12] This is potentially advantageous
as it allows processing of heat sensitive drugs in the melt at low temperatures. [6] On
the other hand it might also be problematic as it leads to a decreased stability within
the dispersions. Also the choice of stabilizer influences both parameters for example,
saturated phospholipids which increase the crystallization temperature and slow down
the polymorphic transitions. [8]
Particle morphology
The morphology of lipid nanoparticles depends on the composition of the dispersion
2
CHAPTER 1. INTRODUCTION
and polymorphic form of the individual nanocrystal. [6] In the case of triglyceride nanoparticles in the stable β-modification the morphology is typically an edged platelet with
the triglyceride molecules arranged in layers being oriented parallel to the large 001
surface of the platelet. [14–17] The α-modifications are difficult to study due their short
lifetimes. But it was found that stabilization through surfactants can extended their
lifetime as for example, stabilization with saturated lecithin and sodium glycocholate
allows a higher stability of the α-phase and a more detailed study of their structure.
It was found that the particles in the α-phase are less anisometric than those in the
β-phase and that the particles become almost spherical. [6,16] It should also be taken into
account that other colloidal structures might be present in the dispersions studied as the
stabilizers may form additional structures e.g. mixed micelles or vesicles. [6]
Melting behaviour
With decreasing size an unusual phenomena can be observed upon heating which is
usually studied by Differential scanning calorimetry (DSC). A broadening of the melting event is observed in the measured curves and the discrete melting point is transformed into multiple discrete melting events below the bulk melting temperature. As
demonstrated by temperature resolved SAXS studies, the different melting transitions
correspond to the melting of particle fractions of platelets having different thicknesses.
This can be ascribed to the platelet like shape and layered structure of the crystalline
triglyceride nanoparticles. [18,19] For pharmaceutical formulations this allows the preparation of drug carriers that release the drug with a well defined temperature range or
a sustained drug release. A disadvantage is that if low-melting-point-triglycerides are
processed into too small particles they may have melting temperatures below body temperature and therefore loose the advantage of a solid core. Furthermore, if just a fraction
of the particles melts stability problems would occur due to repeated melting and recrystallization. [3] Therefore the idea is to separate particle fractions with similar sizes by
for example, centrifugation to avoid instabilities such as particle growth, gel formation
and the expulsion of incorporated drugs.
Pharmaceutical suitability
With regard to pharmaceutical demands on colloidal dispersions, some requirements
have to be considered. Colloidal dispersions should be composed of biodegradable and
nontoxic components with a low or no reticuloendothelial system activity. They should
not contain a significant fraction of microparticles as this would lead to embolism. The
dispersions should be sterile and have a shelf life of more than three years.
3
CHAPTER 1. INTRODUCTION
However, despite these requirements comparatively little is known about the type of
interaction between drugs and lipid nanoparticles. Due to their anisometric shape and
large specific interface, lipid nanoparticles are promising candidates for high drug load by
adsorption of molecules from the aqueous phase. [6] Therefore the drug incorporation of a
variety of different substance with lipophilic properties into the solid core, for example,
cytostatics, immunosuppressants and liphophilic vitamins, has been studied, but usually
the incorporation capacity was quite low. One reason is the crystalline nature of the
particles as the crystalline matrix has limited space for the incorporation of a foreign
substance. [2,6]
For some drugs (ubidecarenone and ciclosporin) distinctly higher drug loads than commonly observed have been reported. [12,20] But in both cases the drug does not necessarily
seem to be located in the core and strongly influence the physicochemical properties of
the particles. As the incorporation into the core seems to be unfavorable it is assumed
that a large fraction of the drug is frequently localized at the surface of the particles. [6]
This concept has been tested as for example, the adsorption of nucleic acids onto the
surface of lipid nanoparticles with a positive surface charge and peptide adsorption on
the surface of lipid nanoparticles coated with chitosan were studied. [21,22]
Dispersions of triglyceride nanoparticles
Stable liquid dispersions can be prepared with comparatively high nanoparticle concentrations (typically around 10%) . Higher concentrations lead to high viscosity and a
semi-solid gel-like consistency, which can be reversed by dilution or for phospholipidcontaining dispersions circumvented by adding highly mobile ionic or non ionic coemulsifying agents. [11,23,24] Dispersions of triglyceride nanocrystals with concentrations
of c ≥ 4 wt % exhibit a self assembly of the platelets into stacked lamellae. Accordingly SAXS patterns exhibit interferences at small scattering angles wich are roughly
equidistant on a Q-Scale 1 . Besides other parameters the mean interparticle distance
can be estimated from these interferences. If it is possible to stabilize such stacked
lamellae formulations they can possibly be used as a new type of carrier system where
the drug is intercalated in the interparticle gaps. [26,27]
Previous work on tripalmitin dispersions as an injectable, colloidal carrier systems were
carried out by Illing et al.. [28] Deoxyribonucleic acid (DNA) was used as model drug
1
Q is the absolute value of the scattering vector Q which is defined to be the difference vector of the
wave vectors ki and kf of the incoming and scattered wave, respectively. [25]
4
CHAPTER 1. INTRODUCTION
due to its polyanionic characteristics and the high potential for the resulting formulations
in gene therapy.
In the study of Illing et al. [28] , dispersions consisting of tripalmitin, the stabilizers
(S100, a phospholipid mixture, and Poloxamer 188, a polymeric stabilizer), dimethyldioctadecylammonium bromide (DDAB) as cationic surfactant and a water-glycerol mixture were prepared. The ratios were varied to obtain stable dispersions within the desired
size range and polydispersity. The dispersions displayed the characteristic melting behaviour with multiple melting events of the β-phase in DSC experiments. Wide angle
X-ray scattering (WAXS) was used to confirm the crystallisation into the stable β-phase.
Particle self-assembly was observed and confirmed by cryo transmission electron microscopy (Cryo-TEM) and SAXS. [26]
In a second step the preparation of DNA-triglyceride nanoparticle complexes was studied.
DNA-tripalmitin-complexes were obtained by adding small amounts of the tripalmitin
dispersion to the aqueous DNA-solution. The composition of the samples was determined
by the charge ratio (CR) between the positive charge of the cationic surfactant (DDAB)
and the negative charge of the anionic DNA. The complex size was found to strongly
depend on the charge ratio. Cryo-TEM and SAXS measurements confirmed a stacklike assembly of particles. However, the presence of DNA could not be proven. The
DNA-triglyceride mixtures displayed a different melting behaviour compared to the pure
dispersions. The multiple discrete melting events narrowed into a single melting peak
with increasing DNA content of the sample.
1.2 Objective
This work contributes to studies which aim to find a way to formulate DNA in a controlled way to be located between the platelet like shaped triglyceride nanoparticles
which should self-assemble into small well defined stacks dispersed in an aqueous medium.
This thesis is especially focused on the preparation and characterization of trimyristin
nanosuspensions where the nanoparticles are functionalised by a cationic surfactant and
further stabilized by the phospholipide blend Lipoid S100. The desired size of the
particles ranges between 100 nm and 200 nm. In comparison to the dispersions studied by Illing et al. [28] no Poloxamer should be used as co-stabilizer.
5
CHAPTER 1. INTRODUCTION
In order to find a well suited dispersion for a DNA formulation seven different cationic
surfactants were studied with regard to their influence on the trimyristin particles. These
include dimethyldioctadecylammonium bromide (DDAB), 1,2-dioleoyl-3-trimethylammonium-propane chloride (DOTAP), cetylpyridinium (hexadecylpyridinium chloride
monohydrat) (CPY), N-cetyl-N,N,N-trimethylammonium bromide (CTAB), dodecyltrimethylammonium bromide (D2TAB), decyltrimethylammonium bromide (DTAB) and
n-hexyltrimethylammonium bromide (HTAB). The influence of other surfactants on the
crystallisation and polymorphism of lipid nanoparticles is reported in literature. [11,24]
All surfactants are cationic quaternary ammonium compounds. They differ in their
head group and the length of the hydrophilic carbon chain (Fig. 1.2). Moreover DOTAP and DDAB (both C18) possess two hydrophilic carbon chains as opposed to the
other surfactants which have only one chain. The hydrophilic carbon chain of DOTAP
possesses double bonds and thus is an unsaturated lipid, whereas DDAB is a saturated
lipid without double bonds. CTAB, D2TAB, DTAB, HTAB have the same head group
and differ only in their chain length. CPY has the same chain length as CTAB but has
a different head group. Through the positive charge of all surfactants a positive charge
should be introduced onto the surface of the particles as the surfactants stabilize the
solid trimyristin core and form a positive charged shell.
Two different preparation methods, high pressure melt homogenisation and ultrasonication were used and compared. The particles were characterized by PCS, DSC, SAXS
and WAXS and their properties compared to literature results of similar compounds. [28]
Furthermore, DNA-triglyceride nanoparticle complexes were prepared by adding DNA
solution to the previously prepared trimyristin dispersions in order to prepare defined,
stable complexes and to characterize them.
6
CHAPTER 1. INTRODUCTION
Cl-
N
O
O
N+
Br-
O
O
N+ Cl-
N+ Br-
N+ BrN+
Br-
N+ Br-
(a)
(b)
(c)
(d)
(e)
(f)
(g)
Figure 1.2: Schematic representation of cationic surfactants which have been used as
stabilizing agents: (a) dimethyldioctadecylammonium bromide (DDAB,
C18) (b) 1,2-dioleoyl-3-trimethylammonium-propane chloride (DOTAP,
C18) (c) cetylpyridinium (CPY, C16) (d) N-cetyl-N,N,N-trimethylammonium bromide (CTAB, C16) (e) dodecyltrimethylammonium bromide
(D2TAB, C12) (f) dodecyltrimethylammonium bromide (DTAB, C10) (g)
n-hexyltrimethylammonium bromide (HTAB, C6).
7
2 Theory
2.1 Differential scanning calorimetry (DSC)
Many chemical reactions and physical transitions involve the generation or consumption
of heat and the objective of calorimetry is the measurement of heat. Therefore, it is
useful to investigate such processes and by Differential Scanning Calorimetry (DSC) the
heat of a reaction of a process can be measured.
There are two different types of DSC instruments: the heat flux DSC and the power
compensated DSC. For both methods the sample is filled into a sample holder (crucible)
which is subjected to a predefined temperature program. The schematic set-up of both
instrument types is displayed in Fig. 2.1. The heat flux DSC has just one furnace for both
the sample and the reference crucibles and measures the heat flow into and out of the
sample. The power compensated DSC has an individual heater for each sample holder.
The temperature of both samples is kept constant during the temperature program by
instantaneous regulation of the two heaters. From the applied heating power of the two
heaters the heat consumption of the sample with respect to the reference sample per
time unit can be extracted directly.
After the adequate calibration any reaction of a material with an evolution of heat can
be studied by both methods with no significant differences and characteristic temperatures such as melting points but also the enthalpy of the corresponding reaction ∆f us H
can extracted from the measured data. There are various material properties that can
be deduced by DSC such as the temperatures and specific heats of first and second order
transitions. An example for a first order transition is the melting of a crystalline compound from the ordered solid state to the disordered liquid or molten state. [29] Other
first order transitions are crystallization and polymorphic transitions. However it should
be kept in mind, that even though DSC provides a detailed analysis of the generated
heat it does not reveal the direct cause of a thermal event.
8
CHAPTER 2. THEORY
(a)
(b)
Figure 2.1: Schematic representations of a) power compensated differential scanning
calorimeter and b) a heat flux differential scanning calorimeter. [29]
Figure 2.2: DSC heating scan of sucrose (undried) with a heat flux DSC, showing the
glass transition temperature (Tg ), recrystallisation temperature (Tc ) with
enthalpy (∆Hc ), melting temperature (Tm ) with enthalpy (∆Hf ) and the
onset of degradation at a heating rate of 10 K min−1 . [29]
9
CHAPTER 2. THEORY
The signal obtained for the heat flow is usually analyzed as a function of time or temperature. A typical DSC measurement of a heat flux DSC is shown in Fig. 2.2. [29] The
integration of the signal corresponds to the heat exchanged during the transition and
even if the melting events look distorted due to instrumental limitations, the overall
enthalpy is unaffected and ∆f us H can be calculated. When interpreting the obtained
signals it should be considered that the material itself also influences the peak shape as,
for example, pure bulk materials produce a ”neat” signal whereas dispersed materials
broaden the shape of the signal.
The scanning rate effects the peak resolution and sensitivity of the instrument as it
determines the heat flow into and out of the sample. Slow scan rates are preferred when a
good peak resolution is required as the sample has a longer time to reach equilibrium and
the transformation takes place over a long time range. Whereas high scan rates increase
the sensitivity of the measurement as the heat exchange takes place in a comparatively
short time. The applied scanning rates depend on the instrument used and can range
from 0.001 to 10 ○C/min. [25] It is important to use the same scanning rate for calibration
and the measurement.
For calibration of the temperature scale but also the enthalpy scale a reference substance
of high purity [30] with a well defined first-order phase transition [31] is subjected to a calibration run. The temperature scale is calibrated by measuring the melting temperature
of two substances and the precalibrated temperature sensors of the instrument are accordingly fine tuned. This procedure is applied correspondingly for the enthalpy of heat
and heat capacity. The aim is to establish a relationship between values of a quantity
indicated by a measuring instrument or measuring system and the corresponsing values
realised by standarts. [31] The most common calibration substance is indium (mp 156 ○C)
which is suitable for temperature as well as for heat calibration. Other commonly used
reference materials used for calibration are tin, lead or zinc. These substances, which
cover a broad temperature range, are rather unsuitable when working with organic material. Therefore some other substances such as cyclopentane or naphthalene should be
included in the calibration process. [25]
The DSC used in this project is a heat flux µ-DSC from Setaram which allows measurements at very low heating rates of 0.001 K min−1 to 1 K min−1 . A temperature range of
−20 ○C to 120 ○C is covered, but actual measurements were performed from 6 ○C to 60 ○C.
The instrument was calibrated with naphthalene. Furthermore, a power compensated
DSC from Perkin Elmer was used to measure at faster scanning rates of 10 K min−1 and a
10
CHAPTER 2. THEORY
temperature range between 0 ○C and 80 ○C. The power compensated DCS was calibrated
with indium.
Triglyceride nanoparticle dispersions require special care concerning the sample preparation as for example, dilution can alter the physical properties. Measurements in the
native state without dilution and slow scan rates ensure a good resolution of the measurement data and the observation of typical behaviour, as for example, the unusual
melting behaviour and crystallization of the different crystalline phases.
2.2 Small Angle X-Ray-Scattering (SAXS)
Small angle X-ray scattering (SAXS) is a technique that uses X-rays to determine the
structure of materials within a length scale of 1 nm to 1 µm and to study their shape
and size using the principles of diffraction. [32]
The basic principle of the scattering experiment (Fig. 2.3) is that a monochromatic
incoming X-ray beam passes the sample and is scattered by scattering length density (sld)
fluctuations b(I) within the sample. The scattered X-rays are detected as a function of
the scattering angle 2θ. As X-rays are scattered mainly by the electrons in the sample
the scattering length can be expressed by the Thomson scattering length.
Scattering processes are characterized by a reciprocity law, which gives an inverse relationship between the sld fluctuations and the scattering angle. [33] This inverse relationship is already expressed by the well known Bragg’s law:
2d sin θ = λ
(2.1)
Figure 2.3: Schematic representation of a scattering experiment, showing the scattering
triangle. [25]
11
CHAPTER 2. THEORY
where d is the characteristic distance between the lattice plains. It describes the relation
between the scattering angle of a constructive interference of X-rays scattered at a periodic electron density distribution with a periodic distance d. For X-rays the difference
between the particle size of interest (1...1000 nm) and the X-ray wavelength is large
as typical wavelength of X-rays usable for structure analysis are in the range of 1 Å to
6 Å.
The general set-up of a SAXS instrument is simple, but the interpretation of recorded
SAXS data is sometimes ambiguous and without complimentary information about the
sample it is often difficult to interpret the obtained SAXS patters in terms of the structure
of the sample studied.
Advantages of SAXS compared to microscopic methods are the fact that the obtained
information refers to the complete sample volume, the application of in-situ measurements and a simple sample preparation, for example, in the field of biological systems.
Biological samples can also be studied by Cryo-transmission electron microscopy which
requires a complex sample preparation which is prone to the production of artefacts.
With SAXS such material can be studied in its native state without artefacts. Nevertheless Cryo-TEM is indispensable as a complimentary method for biological samples.
The in-situ character of the SAXS method allows studies of samples under pressure,
shear, flow or temperature changes. SAXS measurements can be easily combined with
other methods such as dynamic light scattering (DLS), DSC and ultraviolet-visible spectroscopy (UV-VIS). Nevertheless, microscopy is still commonly more often used though
the sample handling is more difficult because the information gained can be much easier
to access and to analyze compared to SAXS. [34]
2.2.1 General considerations
There are two kinds of scattering, inelastic and elastic scattering. In inelastic scattering
part of the kinetic energy of the incident beam is lost during the scattering process giving
rise to some internal processes. One example is Compton scattering where a photon is
scattered at a charged particle and the wavelength changes.
For the description of SAXS patterns we assume that only elastic scattering occurs
meaning that the wavelength of the incident beam is not changed during the scattering
process. [32] Inelastic scattering can be neglected. The incident beam is described as a
plane wave with the wave vector ki and the scattered beam by a plane like wave vector
12
CHAPTER 2. THEORY
kf (Far field approximation). The scattering vector Q is defined by the difference of the
incident and scattered wave vectors:
Q = kf − ki
(2.2)
As mentioned above, only the elastic case ∣kf ∣ = ∣kf ∣ = 2π
λ (λ: wavelength) is considered
and from the scattering triangle (Fig. 2.3) the magnitude of Q is related to the scattering
angle 2θ via
4π sin( 2θ
2 )
(2.3)
Q = 2πs =
λ
For typical SAXS measurements thin samples with transparencies of 90% with respect
to scattering are used. Correspondingly kinematic scattering theory can be used for the
calculation and interpretation of SAXS patterns respectively. Multiple scattering effects
can also be neglected.
Within this approximation the scattered intensity I(Q) can be described by the electrical
field vector E of the scattered wave:
I(Q) = ∣E(Q)∣
(2.4)
In the far field approximation the scattered wave can be shown to be related to the spatial
distribution of the electron density of the sample structure by a Fourier transformation:
E(Q) =
e2 sin θz
E0 ∫ ρ(r)eirQ dV
me c2 R
(2.5)
where e is the elementary charge, me the electron mass, c the velocity of light in a
vacuum and θz the polarisation angle with respect to z-direction when the incoming
beam is polarised to z-direction.
For a discrete distribution of the electrons in the sample Eq. 2.5 can also be rewritten
as the sum over the scattered waves of all electrons:
n
e2 sin θz
E
eire,j Q
∑
0
me c2 R
j=1
(2.6)
n n
e4 sin2 θz
2
∣E
∣
⋅
∑
∑ cos [(re,j − re,k ) ⋅ Q]
0
m2e c4 R2
k=1 j=1
(2.7)
E(Q) =
And I(Q) reads as:
I(Q) =
Here re,j is the postion vector of the jth atom and re,k the position vectors of the scattered
wave.
13
CHAPTER 2. THEORY
Figure 2.4: The visual description of the position vector re which is a the sum of several
vectors to describe an arbitrary position inside a nanocrystal. [25]
2.2.2 Scattering of a dispersion of lipid nanocrystals
The kinematic scattering theory can also be applied to triglyceride nanoparticles, but due
to their crystalline structure the calculation of the patterns become rather complicated,
in particular at high concentrations where locale texture of the particles orientation
occurs. Based on the kinematic scattering theory a model for the ab initio calculation
of the scattering pattern was developed. [35] Triglyceride nanoparticles are crystalline
and platelet-like shaped in their stable β-modification which can be approximately be
described by a parallelepipedic shape.
Instantaneous position of an electron is described by a position vector re within such
a parallelepipedic crystal (Fig. 2.4) and can be written as the sum of vectors in the
following form:
3
̃
re = Rk + ∑ mi ai + rj + R
(2.8)
i=1
where Rk is the point of origin of the coordinate system of a particular nanocrystal k,
the sum over mi ai describes the position of a particular unit cell, rj is the position of
̃ points to the infinitesimal volume element within
the atom j within the unit cell and R
the jth atom [35] .
Inserting Eq. 2.8 in Eq. 2.5 gives :
N k −1 N k −1 N k −1
Nk 1
NA
2
3
e2 sin θz
′ ir Q j
E
E(Q) =
∑
∑
∑
∑
∑ ∫ j E e e ρe dV.
0
2
me c R
k=1 m1 =0 m2 =0 m3 =0 j=1 VA
14
(2.9)
CHAPTER 2. THEORY
where E0 is the amplitude of the incoming wave and ρje the electron density of the jth
atom which is integrated over the volume of the jth atom VAj to calculate the scattering
contribution of the jth atom in a unit cell. E ′ eire Q describes the scattered wave. NA is
the number of atoms in a unit cell, Nik is the number of unit cell planes in the direction
i of crystal k and Nk the number of crystals in the respected coherence volume.
The summation of the scattering contributions of all unit cells and atoms can be rewritten
to:
Nk
k
E(Q) = Ef0 ⋅ ∑ {eiRk Q ⋅ (F k (Q) ⋅ Gk (Q) − SD
(Q))}
(2.10)
k=1
with Ef0 as the amplitude of the scattered wave, the structure amplitude F k (which
includes the scattering of all atoms of the unit cell and ∣F ∣2 is the intensity of the
Bragg peak), the lattice amplitude Gk (interference of periodically arranged unit cells;
k
describes the position of the Bragg peak) and SD
which includes the particle shape and
shell properties.
By multiplication of E(Q) with its conjugated complex function the intensity scattered
by a coherence volume of a dispersion of triglyceride nanoparticles is obtained :
I(Q) = E(Q) ⋅ (E(Q))∗
(2.11)
The scattering model presented for the description of SAXS of suspensions of organic
nanocrystalls was successfully tested for aqueous dispersions of tripalmitin nanocrystals.
It can be used to fit experimental SAXS-patterns of diluted and native dispersions. It
describes and explains the broadened 001 Bragg reflection including the interparticle
interferences quantitatively with a single set of parameters. It is possible to extract
information about the particle shape, size and their distributions and the arrangement
of the stabiliser molecules in the solid-liquid interface of the particles. [35]
2.3 Photon correlation spectroscopy (PCS)
Photon correlation spectroscopy is a scattering technique used to analyse small particles
within a length scale ranging from 5 nm to 5 µm. [36] It is also known as quasi elastic or
dynamic light scattering [37] [38] and it is possible to obtain information on the particle
size and its polydispersity expressed as a polydispersity index (PI). This technique uses
the phenomenon of light scattering to analyze particles in a fluid and has the advantage
15
CHAPTER 2. THEORY
being non-destructive and requiring only small amounts of sample. In practise, the
method measured the intensity fluctuations of the light scattered from a laser beam
passing through the sample volume.
Particles in solution perform a thermally induced diffusion also known as Brownian motion. [39] The Brownian motion can mathematically be described by the random walk
model. The measured intensity fluctuates in time due to interference of the light
scattered by the moving particles. The fluctuations causes a time and place dependent
autocorrelation function G(r, t).The dynamic structure amplitude F (q, t) gives information on the particle motion and forms a Fourier Pair with G(r, t), which means they
can be transformed into each other by Fourier transformation, respectively:
Fs (q, t) = ∫ Gs (r, t)exp(iqr)dr
(2.12)
and
N
N
(0, 0) (r, t)⟩
(2.13)
V
V
Here q is the scattering vector, r the relative distance vector of the scattering particle
and N
V gives the particle density. The functions are averaged to describe the whole
scattering volume and the total measuring time ⟨. . .⟩.
Gs (r, t) describes the probability to find a particle at time t and position r, if the
same particle was previously located at 0 for time and position. The Brownian motion
described by the random walk model Gs (r, t) is:
Gs (r, t) = ⟨
Gs (r, t) = [
3/2
2π
3r(t)2
< ∆R(t)2 >] exp(−
)
3
2 < ∆R(t)2 >
(2.14)
where ∆R(t)2 is the mean squared displacement of the scattered particles. A Fourier
transform leads to:
Fs (q, t) = exp(−q 2 < ∆R(t)2 > t/6) = e−Dq
2t
(2.15)
The Stokes-Einstein-Equation is:
D=
kT
kT
=
f
6 π ηLM Rh
(2.16)
where k is the Boltzman constant, η the dynamic solvent viscosity and Rh is the hydrodynamic radius. RH can be determined if η and the sample temperature T are known
and provides a good estimation of the particle size under the assumption that the particle
is spherical.
16
CHAPTER 2. THEORY
For dynamic light scattering a detailed analysis of the fluctuating intensity is important
and the fluctuation patterns are transferred into an intensity auto correlation function
g2 (τ ) where the time dependent scattered Intensity I is multiplied with itself shifted by
distance τ in time and the product is averaged over the whole measurement time:
g2 (τ ) = ⟨I(q, t) ⋅ I(q, t + τ )⟩
(2.17)
The intensity should decay exponentially and if it is related to the dynamic structure
amplitude Fs (q, t), one obtains the following equation, also known as Siegert relation:
√
⟨I(t) ⋅ I(t + τ )⟩ − A
2
F (q, τ ) = exp(−Dq τ ) = ⟨E(q, t)E(q, t + τ )⟩ =
(2.18)
A
or
√
F (q, t) = g1 (q, t) =
G2 (q, t) − A
A
(2.19)
with A = ⟨I⟩2 as the base line of correlation function.
Polydisperse samples which have a size distribution P (R) have a corresponding distribution function of the self-diffusion coefficients P (Ds ) which is instead of a monoexponential decay a superposition of several exponential functions:
Fs (q, τ ) = ∫
∞
0
P (Ds )exp(−q 2 Ds τ )dDs
(2.20)
In practise, the function is analysed by the ”cumulant” analysis, which is a series expansion of Fs (q, τ ) and only valöid for small size polydispersities (<20%):
lnFs (q, τ ) = −κ1 τ +
1
1
κ2 τ 2 − κ3 τ 3 + ...
2!
3!
(2.21)
From the first cumulant κ1 = D̄s q 2 the average diffusion coefficient D̄s can be obtained
and therefore RH as well. The polydispersity of diffusion coefficients σD is provided by
2
the second cumulant κ2 = (D̄s2 − D̄s )q 4 :
√
2
√
D̄s2 − D̄s
κ2
σD =
(2.22)
=
κ21
D̄s
The polydispersity index (PI) give a rough estimation on the particle size distribution
with some general values: a PI between 0.03 and 0.06 can be considered as a monodisperse solution with all particles having more or less the same size. A PI between 0.1
and 0.2 is a broader but still rather narrow distribution and values between 0.25 and
0.5 indicate a broad distribution and a polydisperse sample. PI values above 0.5 lead to
spurious results.
17
CHAPTER 2. THEORY
Figure 2.5: Schematic representation of the ζ-potential. [41]
2.4 ζ-Potential
There are many different types of colloidal suspensions with different physical properties
and behaviour. Their characterization and control is desirable as it can improve the
performance of the dispersions. The surface potential and the agglomeration tendency
are characteristic properties which can be expressed by the ζ-potential.
Dispersion properties can be influenced by surface forces which increase as the particle
size decreases and the specific surface expands. [40] Most colloids in aqueous solutions
posses an electrical surface charge causing electrostatic repulsion between adjacent particles and the formation of the so called double layer. The double layer is a model used
to describe the arrangement of ions at the solid-liquid interface (Fig. 2.5).
The liquid layer surrounding a charged particle consists as two parts: One is a layer of
ions with the opposite charge as the particle firmly attached to the particle surface, the
Stern layer. The radius of the Stern layer is called the radius of shear and is the major
factor determining the mobility of the particles. [42] The other part is a more diffuse
layer containing also ions of the opposite charge as the particle but less attached to the
particle and with a higher mobility. In this layer also ions with the same charge as the
18
CHAPTER 2. THEORY
particle occur and their concentration increases with the distance to the particle due to
repulsion. The ζ-potential is described as the electrical potential at the radius of shear
relative to its value in the distant, bulk medium. [42]
The ζ-potential strongly influences the interaction among colloidal particles and is consequently related to the stability of a colloidal system. Depending on the value of the
ζ-potential, the tendency of the particles to repel each other is higher or lower.
The DLVO-theory (Derjaguin, Verwey, Landau and Overbeek theory) describes the summation of all attractive and repulsive forces and explains the stability of colloids. The
combination of the electrostatic repulsion and the Van der Waals- attraction forms the
net interaction energy curve which describes the stability of a colloidal system and gives
indication on its alteration. Possible modifications include a variation in pH, the ionic
strength or the addition of surface active agents.
By changing the pH or ionic strength, the charge inside a colloidal system changes thus
causing either an improved or reduced stability. In general, a particle with a ζ-potential
higher than +30 mV or lower than -30 mV is considered stable. [41]
19
3 Experimental section
3.1 Materials
Trimyristin (TM; 95 %) was donated by Sasol, 58453 Witten, Germany. S100 is purified
soybean lecithin, 94 % phosphatidylcholine (PC) (Tab. 3.1). It consists mainly of phospholipides with unsaturated fatty acids. S100 and 1,2-dioleoyl-3-trimethylammoniumpropane chloride (DOTAP) were donated by Lipoid, 67065 Ludwigshafen, Germany.
Poloxamer 188 (PX) also known as Pluronic F-68 is a Polyoxyethylene-polyoxypropylene
block copolymer. Poloxamer 188, cetylpyridinium chloride (hexadecylpyridinium chloride monohydrat) (CPY) (≥ 99.9 %), dimethyldioctadecylammonium bromide (DDAB)
(≥ 98.0 %) and deoxyribonucleic acid sodium salt from hering testes, Type XIV (6.5 %
sodium) were purchased from Sigma, 89555 Steinheim, Germany. N-cetyl-N,N,N-trimethylammonium bromide (CTAB) was purchased from Merck, 64293 Darmstadt, Germany.
Dodecyltrimethylammonium bromide (D2TAB), decyltrimethylammonium bromide (≥ 99.9 %)
(DTAB) and n-hexyltrimethylammonium bromide (> 98 %) (HTAB) were both purchased from TCI, 65760 Eschborn, Germany.
All chemicals were used without further purification.
Milli-Q-water with a specific electrical resistance of ρ>18.2 MΩ⋅cm was used in all experiments.
Fatty acids
chain length : double bonds
Fraction [%]
Palmitic
Stearic
Oleic
Linoleic
Alpha-linolenic
16:0
18:0
18:1
18:2
18:3
12–17
2–5
11–15
59–70
3–7
Table 3.1: Fatty acid composition of S100 normalized to the total number of fatty acids. In average
86% of the fatty acids are C18 chains, about 83% possess at least one unsaturated bond
(18:x with x > 0).
20
CHAPTER 3. EXPERIMENTAL SECTION
3.2 Methods
3.2.1 Photon correlation spectroscopy measurements (PCS)
The particle size, which provides a good estimate for the average diameter of the platelet,
and the polydispersity index were determined with the cumulant analysis of the correlation function measured by a photon correlation spectrometer (Brookhaven Instruments
Corporation, 11742 Holtsville, NY, USA) comprising of a Mini-L 30 compact diode laser
(30 mW,637 nm) and a BI-200 SM goniometer carrying the photo multiplier tube. The
goniometer was fixed at an 90° angle with respect to the incident laser beam. An entrance slit of 100 µm and a wavelength filter of 633 nm were inserted in front of the photo
multiplier.
A few droplets of the dispersion were added to a glass cuvette and diluted with Milli-Q
water without further filtration until an appropriate counting rate of 50 to 100 kcps was
obtained. The samples were measured for 2 min at T = 22 ○C.
3.2.2 Small and wide angle X-ray scattering measurements (SAXS and
WAXS )
SAXS and WAXS measurements of diluted trimyristin dispersions were performed using
a Kratky-type camera (S3-MICROpix, Hecus X-Ray Systems, 8020 Graz, Austria) of
the Hard-Soft-Matter lab at the FRM II of the TU München in Garching.
The source comprises a 50 W Cu Kα X-ray source and a bi-ellipsoidal FOX 3D graded
W/Si multilayer mirror optics (both from Xenocs, 38360 Sassenage, France), selecting
the Cu Kα wavelength of 1.5418 Å at a flux of about 3⋅108 cps . The mirror optics focuses
the primary beam onto the detector. The Kratky block collimator possesses 200 µm and
1000 µm slits in the vertical and horizontal directions, providing a beam size of about
0.2 mm x 0.25 mm at the sample position and a flux of about 107 cps, as measured with
a pin diode. To reduce background scattering due to air scattering the beam path in the
camera housing is kept under vacuum (2 mbar).
Two-dimensional SAXS patterns were recorded with a Pilatus 100K detector (Dectris
AG, 5400 Baden, Switzerland) at 22 ○C. The detector allows direct detection of X-rays
and single-photon counting and provides an excellent signal-to-noise ratio (zero dark
current and read-out noise) and a very high dynamic range (20 bit). For the detection
21
CHAPTER 3. EXPERIMENTAL SECTION
of the WAXS signal a Mythen 1K line detector (Dectris AG) with 1280 channels and
based on the same technology as the Pilatus detector was used.
The dispersions were measured in a quartz capillary (Hilgenberg GmbH, 34323 Malsfeld,
Germany) with a mean diameter of 1 mm and a wall thickness of 10 µm. The capillary
was glued into a custom build stainless steel holder which fixes the position of the sample
in the beam. The capillary holder can be sealed vacuum-tight with screw caps. Using
the same capillary and same sample position allows constant measurement conditions,
facilitating background subtraction even for weakly scattering samples like the diluted
trimyristin dispersions. The temperature of the sample stage housing the capillary holder
was set for all measurement to 22 ○C.
The sample-detector distance was calibrated to 289.5 mm using silver behenate (Eastman
Kodak Company) with a long spacing of 58.38 Å as a standart sample. [43] Transmission
of each sample was measured for 0.1 s with the Pilatus detector with the W-beamstop
beeing removed. The transmission was determined from the ratio of the measured intensity and the initial beam intensity. The real thickness of the capillary was calculated
via transmission runs for an empty capillary and a capillary being filled with D2 O to
be 0.76 mm. [44] Calibration of the scattering intensity to an absolute scale was performed with a glassy carbon sample [45] provided by the 15ID-D USAXS beamline at the
Advanced Photon Source, 60439 Argonne, IL, USA. Using the calibration factor, the
transmission for each sample and the thickness of the capillary, all scattering curves can
be normalised to an absolute scale. [46]
Data reduction using the one-dimensional scattering law (azimuthal average, absolute
scale) was performed using fit2dcorr, a C++-extension for fit2d. [47,48] The scattering
pattern of H2 O was subsequently subtracted from the sample patterns. The WAXS data
was calibrated using pure tripalmitin powder, reference values for the peak positions were
taken from the A2 beamline of the Hasylab in Hamburg [49] . As for SAXS, the WAXS
pattern of H2 O was subtracted from the sample curves.
The samples were measured in their native form (10 wt% trimyristin) and as well in their
diluted form with up to 3 wt% trimyristin including DNA. DNA loaded samples were
prepared immediately before measurement.
22
CHAPTER 3. EXPERIMENTAL SECTION
3.2.3 DSC measurements
µ-DSC measurements were carried out using a Micro DSC III instrument (Setaram,
69300 Caluire-et-Cuire, France). The samples were heated at a scan rate of 0.1 ○C/min
from 6 ○C to 60 ○C and subsequently cooled to 6 ○C. About 200 mg of the sample was filled
in a batch cell and the same amount of water was filled into the reference cell. The measurements were performed using the Setaram Calisto Data Aquisition software (v.1.094)
and the resulting data was analysed using Calisto Processing software (v.1.094).
The native trimyristin dispersions and DNA-trimyristin-complexes were studied using a
power compensated DSC instrument (Perkin Elmer, Massachusetts 02451, USA). The
samples were heated at a rate of 10 ○C/min from 0 ○C to 80 ○C and subsequently cooled to
0 ○C. About 10 mg of the sample was filled into an aluminium tin and sealed afterwards.
For measurement and data analysis the software Perkin Elmer ”Pyris” was used.
3.2.4 ζ-Potential measurements
A Zetasizer Nano (Malvern Instruments) equipped with a He-Ne laser (633 nm, max.
5 mW) was used to measure the ζ-potential of the dispersion. The sample cell was filled
with 2 mL of the diluted sample and a voltage of 150 mV was applied. All measurements
were repeated three times and the results were averaged.
3.3 Preparation of the nanodispersions
In the following section the methods used in the preparation of nanodispersions are described. All compositions of aqueous nanodispersions are given in %wt and summarized
in Tab. 3.2.
3.3.1 Ultrasonication
Two different formulations of an trimyristin dispersions were prepared (NC1 and NC2,
Tab. 3.2). Both formulations contained 10 % trimyristin and 0.4 % of a co-stabilizer.
The first formulation contains additionally 3 % S100 and 4.5 % Poloxamer 188 as costabilizer DDAB was used. The second formulation contains only 2.4 % S100, 0.4 %
23
CHAPTER 3. EXPERIMENTAL SECTION
Figure 3.1: Schematic display of the varied parameter: the amplitude, treatment time
and pulsation cycle.
co-stabilizer and no Poloxamer. Seven different co-stabilizers were used: dimethyldioctadecylammonium bromide (DDAB), cetylpyridinium, (CTAB), Dodecyltrimethylammonium bromide (D2TAB) and decyltrimethylammonium bromide (DTAB), n-hexyltrimethylammonium bromide (HTAB) and 1,2-dioleoyl-3-trimethylammonium-propane
chloride (DODAP).
The phospholipids S100 and trimyristin were heated to 80 ○C until a clear yellowish
melt was obtained. The co-stabilizer was dissolved in H2 O and heated to the same
temperature. The mixture was predispersed for 3 min at 65 ○C with an Ultra-Turrax
T25 Basic disperser (IKA-Werke, 79219 Staufen, Germany) at 22,000 rpm.
The hot emulsion (1 mL) was treated with a Sonopuls ultrasonic homogenizer with a
MS 73 ultrasonic needle (Bandelin electronic, 12207 Berlin, Germany). The ultrasonic
needle is immersed to a maximum depth of 2 cm into the liquid avoiding contact with
the reaction vial. The position of the reaction vial is adjusted before the treatment is
started.
Three different process parameters were varied. These are the amplitude of the sonication, the duration of the treatment and the pulsation time during a 10 s cycle. Each
pulsation cycle has a constant length of 10 s whereas the pulse-on and pulse-off time
vary (Fig. 3.1). For example, cycle 1 has a 1 s pulse-on and 9 s pulse-off time. After
sonication dispersions were allowed to cool to 22 ○C and subsequently to 6 ○C.
24
CHAPTER 3. EXPERIMENTAL SECTION
3.3.2 High pressure melt homogenisation
The preparation of the Poloxamer free dispersions was performed using high pressure
melt homogenisation (HPMH). Two different Poloxamer free formulations were prepared
(NC2 and NC3, Tab. 3.2). Only four different co-stabilizers were used: CPY, CTAB,
D2TAB and DTAB. The first formulation of Poloxamer free dispersions was prepared
with a low co-stabilizer concentration of 0.4 % and the second formulation was prepared
with higher co-stabilizer concentration of 1.2 %.
The pre-emulsion was prepared in the same way as for ultrasonication. The hot preemulsion (60 ml) was passed through a continuous APV-2000 high–pressure melt homogenizer (APV Deutschland, 59425 Unna, Germany) which had been preheated with an
electric heating tape to temperatures above 60 ○C. The dispersions were homogenized
at pressures between 1.0 and 1.5 kbar for 4 minutes. With typical flow rates of 2.5 to 3
mL/s, this corresponds to about 17 to 21 cycles. A previous study had shown that the
particle size does not further decrease after about 16 cycles for this type of homogenizer
and these kinds of dispersions. [50]
The nanoemulsions were allowed to cool to 22 ○C and then to (6 ○C) in order to crystallise.
sample
S100
[%]
Co-stabilizer
[%]
Poloxamer 188
[%]
NC1
3.0
NC2-2
NC2-3
NC2-4
NC2-5
NC3-2
NC3-3
NC3-4
Preparation method
DDAB
0.4
4.5
Ultrasonication
2.4
2.4
2.4
2.4
CPY
CTAB
D2TAB
DTAB
0.4
0.4
0.4
0.4
–
–
–
–
Ultrasonication,
Ultrasonication,
Ultrasonication,
Ultrasonication,
2.4
2.4
2.4
CPY
CTAB
D2TAB
1.2
1.2
1.2
–
–
–
HPMH
HPMH
HPMH
HPMH
HPMH
HPMH
HPMH
Table 3.2: Composition of the dispersions prepared by ultrasonicaction and HPMH.
which all contain 10 wt% trimyristin, and 0.4 wt% co-stabilizer;
Us:Ultrasonication
25
CHAPTER 3. EXPERIMENTAL SECTION
3.3.3 Preparation of DNA-trimyristin nanoparticle-complexes
In addition to the trimyristin dispersions described above, it was attempted to produce
also DNA-trimyristin nanoparticle-complexes. The theoretical charge ratios (CR) of the
complexes were calculated using the following equation:
CR =
mstab /Mstab ⋅ NA ⋅ z
Nstab ⋅ 1
=∣
∣
mDN A /MDN A ⋅ NA ⋅ z
NDN A ⋅ 2
(3.1)
where mstab and Mstab are the mass and molecular weight of cationic co-stabiliser respectively, mDN A and MDN A are the mass and the molecular weight of DNA respectively, NA
is the Avogadro constant and z is the charge of the respective molecule.The co-stabilizer
molecule possesses one positive charge and the DNA two negative charges. Nstab and
NDN A denote the numbers of the corresponding molecules respectively. The amounts of
DNA solution, water and trimyristin dispersion were calculated using a python script (
p.60 in the appendix).
The experiments were conducted for trimyristin dispersions with 0.4 % and 1.2 % costabilizer concentration. Samples with the charge ratios 0.3, 0.5, 0.7, 1 and 2 were
prepared. For comparison a DNA-free sample was prepared.
An aqueous DNA solution (1 wt%) was stirred for approximately 2 h until a clear solution
was obtained. The appropriate volume of DNA solution and the required amount of
water was transferred into a 1.5 mL reaction vial. 0.3 g of the trimyristin dispersion was
first added to the lid of the reaction vial leading to a final trimyristin concentration of
3 %. The lid was closed and directly vortexted for 30 s. Highly viscous samples had to
be shaken by hand to obtain a proper distribution.
26
4 Results and discussion
4.1 Trimyristin nanodispersions prepared by ultrasonication
Triglyceride nanoparticles have been proposed as a new type of drug carrier system
with different pharmaceutical applications, for example intravenous administration. [4,51]
A number of methods for the preparation of triglyceride dispersions have been investigated and developed. For lipid nanoparticles a successful preparation is known since
the beginning of 1990. One of them is the use of high pressure homogenisation which
leads to compositions similar to colloidal fat emulsions and which allows large-scaleproduction. [2]
Tripalmitin dispersions were studied as an injectable, colloidal carrier system and considering DNA loaded dispersions their possible application in gene therapy is discussed. [23,26,28]
The good results obtained, encouraged us to further explore this system with the aim of
improving the preparation methods. This study used trimyristin as the matrix stabilized
by S100 and only a single co-stabilizer belonging to the family of quaternary ammonium
compounds. [28]
To optimize the preparation of these dispersions for particle sizes between 100 nm and
200 nm it was attempted to prepare stable dispersions with a minimum of co-stabilizer
and without the addition of Poloxamer. In addition, two preparation methods were
compared to investigate whether there is a simpler alternative to high pressure melt
homogenisation.
4.1.1 Preparation
As a laboratory method high pressure melt homogenisation (HPMH) is a tedious process
as the assembly and the cleaning of the machine is laborious. There are high pressure
homogenizers available for small sample volumes but for the studies presented here there
27
CHAPTER 4. RESULTS AND DISCUSSION
was just a APV 2000 machine available, prepared such that a minimum of 35 ml sample
is needed.
In contrast, ultrasonication is advantageous for screening processes as it allows fast and
simple preparation of a large number of samples. Ratios and treatment parameters can
be varied easily and thus allow a small scale production which is beneficial with regard
to expensive chemicals and reduction of chemical waste.
For this reasons ultrasonication was chosen as a preparation method in the current study
of trimyristin dispersions in addition to HPMH.
After the preparation of the samples with ultrasonication the samples can be cooled down
to complete the crystallization. The poloxamer containing samples were not cooled down
but it can be assumed that the droplet size is correlated to the platelet size and therefore
the sizes stay in a similar size range. Furthermore, it is known that these samples stay
stable after the cooling step. [26] All poloxamer free samples appeared to be stable at
room temperature (22 ○C) without gelation or foam. After cooling to 6 ○C to complete
the crystallization and to obtain platelet shaped particles, the samples with DDAB,
HTAB and DOTAB were found to be unstable and solidified.
4.1.2 Nanodispersions containing Poloxamer
A series of experiments of a stable dispersion was conducted with the composition of 10 %
TM, 2.4% S100, 0.4% DDAB and 4.5% PX and varying three parameters: the sonication
amplitude, the overall duration of the treatment and the pulsation time during each 10 s
cycle (Tab. 4.1). After preparation the dispersions obtained were not cooled down and
the samples were subsequently analyzed by PCS.
Altogether 26 samples were prepared with 21 stable samples and 5 instable samples.
Instable samples were found for pulsation times of 1 to 5 s per cycle with an amplitude of 30 % and 2 min treatment time and either gelation or formation of foam was
observed. The gelation and foam indicated the instability of the samples and the PCS
measurements did not obtain useful data.
Stable samples were found for longer pulsation times per cycle between 6 and 10 s and
treatment times between 2 and 10 min at amplitudes of 30 % and 50 % but also at a high
amplitude of 70 % with a short treatment time of 2 min and a short pulsation time of
3 s.
28
CHAPTER 4. RESULTS AND DISCUSSION
Amplitude
%
duration
[min]
pulsation time/10 s cycle
[s/ 10 s cycle]
30
30
50
30
50
70
2
2,5,7,10
2,5,7,10
2,5,7,10
2,5,7,10
2
1-9
8
8
9
9
3
Table 4.1: Variation of the amplitude, the time of treatment, the cycle in the first screening process
amplitude
%
duration
[min]
pulsation time during 10 s cycle
[s / 10 s cycle]
eff. diameter
[nm]
30
30
30
30
2
5
7
10
8
8
8
8
157
142
182
185
30
30
30
30
2
5
7
10
9
9
9
9
179
182
181
177
50
50
50
50
2
5
7
10
8
8
8
8
162
189
198
197
50
50
50
50
2
5
7
10
9
9
9
9
188
171
195
195
70
2
3
153
Table 4.2: The effective diameter of the particles (measured by PCS) of the stable
samples obtained in the first screening process in relation to amplitude, duration and cycle characteristics.
29
CHAPTER 4. RESULTS AND DISCUSSION
For pulsation times of 6 and 7 s per cycle the measured particle sizes were larger than
350 nm which was above the desired particle size. For the other stable dispersions obtained approximated mean particle sizes between 140 nm and 200 nm were yielded(Tab.
4.2). The smallest particle sizes were found for 8 s pulsation time per 10 s cycle with an
amplitude of 30 % and overall experiment durations of 2 and 5 min. A similar size was
obtained for 3 s pulsation time per 10 s cycle at an amplitude of 70 % for 2 min. The
increase of the amplitude to 50 % for pulsation times of 8 and 9 s per cycle and a longer
treatment time of 10 min did not yield smaller particles. Therefore these parameters
were not considered further.
4.1.3 Poloxamerfree Nanodispersions
Having shown that dispersions with particles in the size range of 100–200 nm can be
obtained using Poloxamer 188 as stabilizer besides DDAB, it was attempted to produce
similar dispersions without Poloxamer 188. In this experiment CPY, CTAB, D2TAB,
DTAB, HTAB and DOTAP were used in addition to DDAB as co-stabilizer. The treatment parameters chosen (Tab. 4.3) for these tests were those which provided the best
performance in the first set of experiments (Tab. 4.2). Further variations of the parameters were likewise tested. After treatment by ultrasonication the dispersions were
cooled down to 6 ○C and afterwards analyzed by PCS.
The resulting cooled dispersions yielded particles in the size range between 220 and
230 nm. Similar sizes were obtained with CPY (30 % amplitude, 5 min, 9 s pulsation
time per cycle; 30 % amplitude, 5 min, 3 s pulsation time per cycle; 70 % amplitude,
2 and 5 min, 3 s pulsation time per cycle). The smallest particles were obtained with
CTAB (30 % amplitude, 7 min, 8 s pulsation time per cycle) with a size of 196 nm.
Due to time limitations no further variations of parameters were tested. So far the results
indicate that a production of particles smaller than 200 nm is not straight forward.
Comparison of the co-stabilizer showed that the stability and particle sizes are influenced
by the size and molecular structure of the co-stabilizer (Figure 1.2). For co-stabilizers
with carbon chains longer than 16 carbon atoms (DOTAP (C18) and DDAB (C18))
and shorter then 10 (HTAB (C6)) no stable dispersion were obtained, suggesting that
dispersion become unstable if the chains are too long or too short.
Studies on the influence of ionic and non-ionic stabilizers have been reported in the literature. [11,24] It was demonstrated that the crystallization behaviour and polymorphic
30
CHAPTER 4. RESULTS AND DISCUSSION
amplitude
%
duration
[min]
pulsation time/cycle
[s / 10 s cycle]
eff. diameter
[nm]
co-stab.agent
30
5
8
30
7
8
30
5
9
30
2
3
30
5
3
70
2
3
70
5
3
solid
226
221
222
232
solid
solid
254
196
222
205
288
225
244
223
244
229
239
DDAB
CPY
CTAB
D2TAB
DTAB
HTAB
DOTAP
CPY
CTAB
CPY
CTAB
CPY
CTAB
CPY
CTAB
CPY
CTAB
CPY
CTAB
Table 4.3: Results of the second screening with ultrasonication showing the effective
diameter of the particles (measured by PCS) in relation to amplitude, duration
and cycle characteristics. The choice of the selected parameters was based on
the best results of the first screening.
31
CHAPTER 4. RESULTS AND DISCUSSION
transitions as well as the stability of the dispersions obtained are strongly influenced by
the ionic or non-ionic nature of the stabilizer. For example, a combination of non-ionic
surfactants and phospholipid (S100) leads to gelation upon storage, suggesting that gelation might be due to specific interactions between the surfactant and the phospholipid.
Furthermore, the gelation behaviour indicates that these interaction are different for
ionic and non ionic stabilizers and that stable dispersions without gelation can obtained
using a combination of ionic surfactant and phospholipid. [24]
The results obtained in this work complemented the results reported in the literature as
the combination of an ionic stabilizers and phospholipid leads to stable dispersions.
4.2 Trimyristin nanodispersions prepared by high pressure melt
homogenisation (HPMH)
4.2.1 Preparation
High pressure melt homogenisation is a common method to prepare stable dispersions
of small nanoparticles. [26] As stable dispersions without Poloxamer could be prepared
with ultrasonication, the same result was expected using HPMH. Since the preparations
by ultrasonication lead to unstable dispersions for DDAB, HTAB and DOTAP; it was
likewise expected that instability also occurs for HPMH. Thus only CPY, CTAB, D2TAB
and DTAB were used as co-stabilizers.
Two series containing different concentrations of the co-stabilizer were prepared, one
containing 0.4 % co-stabilizer (NC2) and the other containing 1.2 % co-stabilizer (NC3)
(Tab. 4.4). In the formulations with low concentrations CPY, CTAB, D2TAB and DTAB
as co-stabilizer were used whereas in the formulations with higher concentrations CPY,
CTAB and D2TAB were added as co-stabilizer. All of the seven dispersions prepared
were found to be stable and did not show any aggregation after crystallization. The
stability sustained for roughly one month and thereafter small aggregates were observed
in all dispersions.
For the samples having low concentrations of co-stabilizer, particle sizes in the range of
110-140 nm were found. The size decreases with decreasing chain length of the stabilizer
(Tab. 4.4). For samples with high co-stabilizer concentrations particle sizes in the range
of 140-160 nm were obtained. The chain length of the co-stabilizer was not found to
influence the resulting particle size (Tab. 4.4). However with increasing concentration
32
CHAPTER 4. RESULTS AND DISCUSSION
co-stabilizer
%
eff. diameter
[nm]
CPY
CTAB
D2TAB
DTAB
0.4
0.4
0.4
0.4
%
%
%
%
137
130
116
112
CPY
CTAB
D2TAB
1.2 %
1.2 %
1.2 %
150
158
143
Table 4.4: Summary of particle sizes for a low (0.4 %, NC2) and high (1.2 %, NC3)
co-stabilizer concentration prepared with HPMH.
of the stabilizer the particle size increases too as for example particles with a stabilizer
concentration of 0.4 % CTAB are 130 nm and for a higher concentration of 1.2 % CTAB
the particles are 28 nm bigger(158 nm) (Tab. 4.4).
Comparing the homogenized particles with particles produced by ultrasonication shows
that particles obtained by HPMH are much smaller. The particle size difference is
around 50 nm as the particle sizes of particles prepared by HPMH range range between
110-140 nm while the particles obtained by ultrasonication have typically sizes around
200 nm. For high pressure homogenization the liquid is pushed with high pressure
through a narrow gap accelerating the fluid on a short distance to very high velocities.
The high shear stress and cavitation forces disrupt the emulsion droplets into smaller
particles. [5] For ultrasonication a small volume is treated by a focused short and intense
sonication resulting in agitation which disrupts the emulsion droplets. The smaller
particles obtained by HPMH might be explained by a higher energy input and the more
homogeneous distribution of the applied pressure in HPMH compared to the ultrasonication.
A positive surface charge of the particles was introduced by using quaternary ammonium
compounds as stabilizer and to verify the positive surface charge ζ-potential measurements were conducted for dispersions with a low co-stabilizer concentration (0.4 %).
All dispersions exhibited high values above +30 mV for the ζ-potential (Fig. 4.1) which
confirms their stability as colloidal dispersions with ζ-values higher then +30 mV are
considered stable.
33
CHAPTER 4. RESULTS AND DISCUSSION
z e ta p o te n tia l
s iz e
1 3 6
1 4 0
1 4 0
1 3 3
1 2 7 ,3
1 2 4 ,7
1 2 0
1 0 0
1 0 0
8 0 ,2
7 5 ,9
8 0
8 0
5 6 ,9
6 0
6 0
3 8 ,7
4 0
s iz e [n m ]
z e ta p o te n tia l [m V ]
1 2 0
2 0
4 0
2 0
0
0
C P Y
C T A B
D 2 T A B
D T A B
Figure 4.1: Comparison between the size and zeta potential for dispersions with a low
co-stabilizer content of 0.4 %
The comparison of the values of the ζ-potential with the particle size does not demonstrate a obvious relation between them as all particles have a similar size but vary
strongly in their potential. A relation to the chain length of the stabilizing agent is
likewise observed as the positive charge increases with increasing chain length.
It might be assumed that similar or even higher values for the ζ-potential would be
obtained for a higher co-stabilizer content (1.2 %). Unfortunately this could not be
verified as the instrument was not available after the preparation of dispersions with a
high co-stabilizer concentration.
4.2.2 SAXS and WAXS measurements
Further characterization of the native dispersions was performed using SAXS and WAXS.
A broadened 001 Bragg reflection and stack related interferences were commonly observed for high particle concentrations and have also been found in previous experiments
with similar dispersions. [26–28] All dispersions were measured in their native state and a
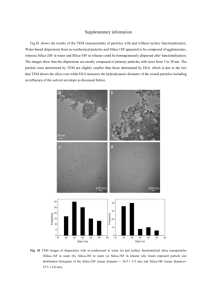
trimyristin concentration of 10 % (Fig. 4.2). For a co-stabilizer concentrations of 0.4 %
and 1.2 % the Bragg reflection was found at the same position independent of the composition of the co-stabilizer (Tab. 4.5). For a high co-stabilizer concentration of 1.2 %
the peak width broadens and is less sharp than for a lower co-stabilizer concentration of
0.4 %. The broadening of the peak indicates that the particle thickness decreases with
34
CHAPTER 4. RESULTS AND DISCUSSION
c o s ta b .
1
0 0 1
3
%
B
A B
B
0 .4
0 .4
0 .4
0 .4
1 .2
1 .2
1 .2
B
A B
In te n s itä t
2
C P Y
C T A
D 2 T
D T A
C P Y
C T A
D 2 T
0 ,1
s [n m
1
-1
]
Figure 4.2: SAXS measurements for native dispersions with 10% of trimyristin. Curves
are shifted in intensity for better visualisation. Arrows mark the stack related
interferences. The numbers mark the order of the corresponding interference
maxima. s is the inverse distance of the lattice planes and I is the Intensity.
35
CHAPTER 4. RESULTS AND DISCUSSION
sample
co-stabilizer
%
s001
[nm]
d001
[nm]
CPY
CTAB
D2TAB
DTAB
0.4
0.4
0.4
0.4
0.276
0.276
0.276
0.276
3.624
3.619
3.619
3.619
CPY
CTAB
D2TAB
1.2
1.2
1.2
0.277
0.277
0.279
3.603
3.603
3.582
Table 4.5: Intensity maximum of the 001 Bragg reflection for the trimyristin dispersions
displayed in Fig.4.2
a higher co-stabilizer concentration as the peak width correlates with the particle thickness. [35,52] As is known from literature [26] stack related interference were observed for
dispersions with a tripalmitin concentration exceeding 4 %. Such stack related interferences were also observed for all dispersions except for the dispersion with a concentration
of 0.4 % DTAB. Interferences up to the third order can be observed but the visibility of
the interferences varies. For 0.4 % CPY, 0.4 % CTAB and the three dispersions with a
high co-stabilizer concentration of 1.2 % the first and the second order can be recognized
and assigned. For a co-stabilier concentration of 0.4 % D2TAB the interferences are less
obvious but they can still be observed. The reflections of the third order were hard to
recognize and were clearly observed only for a low and high concentration of CPY and
D2TAB (Tab. 4.6).
and 2dsinθ = λ the average length d of a repeat unit in a triglyAccording to s = 2sinθ
λ
ceride nanoparticle stack which is the particle thickness plus the interparticle distance
can be calculated. The particle distance within the stacks for the dispersions with 0.4 %
co-stabilizer was about 50 nm and for dispersions with a higher co-stabilizer concentrations it was about 40 nm. Obviously the length of the interparticle repetition unit
decreases with increasing co-stabilizer concentration. This has been reported in literature as well. [26] It was found that the co-stabilizer concentration seems to determine the
distances between the self assembled nanocrystals and a reasonable explanation might
be the enhanced screening of electrostatic replusion due to an increasing ionic strength
in the dispersion medium.
36
CHAPTER 4. RESULTS AND DISCUSSION
s(1st order)
[nm−1 ]
s(2nd order)
[nm−1 ]
s(3nd order)
[nm−1 ]
¯
d(inter
part.dist.)
[nm]
interferences
%
%
%
%
0.0201
0.0186
0,0187
-
0.0405
0.0368
0,0408
-
0.0656
0,0705
-
48.3
54.0
48.4
-
yes
yes
yes
no
CPY, 1.2 %
CTAB, 1.2 %
D2TAB, 1.2 %
0.0249
0.0231
0.0215
0.0520
0.0480
0.0505
0.0837
0.0891
38.2
42.5
40.0
yes
yes
yes
co-stabilizer
CPY,
CTAB,
D2TAB,
DTAB,
0.4
0.4
0.4
0.4
Table 4.6: s-values for the stack related interferences of all 7 dispersions of low costabilizer content (0.4 %) and high co-stabilizer content (1.2 %),respectively
In contrast, the molecular structure of the co-stabilizer did not seem to influence the
distance in the experimental data since all dispersions with different co-stabilizers but
the same concentration have similar d-values.
As mentioned above, the 001 Bragg peak occurred at similar positions for all dispersions
giving similar d-values. In the case of the 001 Bragg reflection the d value is the height
of a trimyristin unit cell parallel to the 001 layer which is vertical to the platelet surface.
The value for bulk trimyristin is d ≈ 3.6 nm. [13] The value obtained for the dispersions
is also as expected d ≈ 3.6 nm and also confirms the triclinic crystal structure.
The WAXS data (Fig. 4.3) gives additional clues about the crystalline modification of
the particles in the dispersions and allows a phase analysis of the triglyceride dispersions.
The WAXS data obtained for the dispersions studied here was compared to synchrotron
WAXS data of tripalmitin which exhibits the same peaks as trimyristin in the wide angle
range (Tab. 4.7). For molecules of a homgeneous series which have a similar structure
and differ in chain length only, it is expected to have similar unit cells and crystal
packing and knowing one member of the series it is possible to predict and determine
the structure of the others. [13] Tripalmitin and trimyristin differ in chain length only
and it is known that both exibit a triclinic crystal structure in the stable β-polymorph
where the molecules are arranged in a tuning fork conformation when crystalline. [9]
The four main peaks (No. 1+2, 6 and 7) could be identified in all seven dispersions (Fig.
4.3) though No. 1 and No. 2 are so close to each other that it is hard to distinguish them.
37
CHAPTER 4. RESULTS AND DISCUSSION
c o s ta
C P Y
C T A B
D 2 T A
D T A B
C P Y
C T A B
D 2 T A
tr ip a lm
1 + 2
1 2 0 0
7
1 0 0 0
6
I [a .u .]
8 0 0
3
5
4
b .
%
0 .4
0 .4
B
0 .4
0 .4
1 .2
1 .2
B
1 .2
itin d iffr a c tio n d a ta
6 0 0
4 0 0
2 0 0
0
2 ,0
2 ,4
2 ,8
s [n m
-1
]
Figure 4.3: WAXS measurements for native dispersions with 10 % trimyristin compared
with synchrotron power diffraction data of tripalmitin from the beam line
A2 Hasylab. The curves are shifted vertically for better visualization.
No. of peak
2 θ for λ=0.15
s (lit.)
[nm−1 ]
s (exp.)
[nm−1 ]
1
2
3
4
5
6
7
18.822
18.968
19.356
20.797
21.591
22.572
23.537
2.1802
2.1969
2.2415
2.4066
2.4974
2.6094
2.7195
2.1679
2.1841
2.2418
2.4054
2.4933
2.5938
2.7067
Table 4.7: Comparision of synchotron powder diffraction data of tripalmitin powder from
beam line A2 Hasylab with the experimental WAXS data of native trimyristin
dispersions.
38
CHAPTER 4. RESULTS AND DISCUSSION
All other peaks disappeared due to background noise. Possibly there are also scattering
contributions of the α phase, but as no distinct peak for the α-phase (sα =2.4 nm−1 )
was observed only a very small amount of this phase might be present which could
not be detected. Hence it is concluded that the dispersions mainly crystallize in the β
modification.
4.2.3 DSC measurements
To obtain information on the melting and crystallization behaviour the stable native
trimyristin dispersions were studied by DSC (Fig. 4.4). Two major endothermal regiemes in the heating curves were observed. The first regime is characterized by a broad
endothermal peak between 27–33 ○C. The second regime exhibits multiple discrete melting events between 35–56 ○C (Fig. 4.4(a)). For bulk trimyristin melting temperatures
for the α- and the β-modification were found to be 33 °C and 56 °C respectively. [10]
The observed melting events agree well with the literature values (Tab. 4.8) allowing to
interpret the first melting regime by the melting of the α-phase and the second by the
melting of the β modification. The multiple discrete melting events in the second regime
are ascribed to the melting of particle fractions of trimyristin platelets having different
thicknesses.
The dispersions displayed differences in the proportion of the α-phase as reflected in the
intensity and enthalpy of the first melting regime. Dispersions with CPY or CTAB seem
to contain a larger proportion of the α-phase than dispersions with D2TAB and DTAB
Comparing the experimental melting temperatures which are between 32 °C and 34 °C,
to the bulk melting temperature of the α-phase (33 °C) no significant difference for both
co-stabilizer concentrations, 0.4 % and 1.2 % is observed.
The enthalpy of fusion ∆Hf us describes the change of enthalpy during a melting process
and indicates how much energy is needed for the melting process. The enthalpy change is
the area under the curve of the heat capacity against the temperature. [42] It is obtained
by integration of the area under the melting curves of both regimes over the temperature.
The experimental values for the enthalpy of fusion range from 14–16 J g−1 which is in fair
agreement with the literature value (17 J g−1 ) for dispersed trimyristin particles. [10]
For the crystallization a single sharp peak is observed for all dispersions studied and the
determined crystallization temperatures corresponds to the peak maxima.
39
CHAPTER 4. RESULTS AND DISCUSSION
a-phase
b-phase
(a)
(b)
Figure 4.4: µ-DSC measurement of stable native dispersions with a high and low co-stabilizer concentration a) heating curves (0.1 ○C/min). Curves are shifted on the y scale for better
visualization b) cooling curves (0.1 ○C/min)
40
CHAPTER 4. RESULTS AND DISCUSSION
trimyristin (bulk)*
trimyristin
(dispersion, 10 %wt) *+
CPY, 0.4 %
CTAB, 0.4 %
D2TAB, 0.4 %
DTAB, 0.4 %
CPY, 1.2 %
CTAB, 1.2 %
D2TAB, 1.2 %
α
Tm
[○C]
β
Tm
[○C]
∆Hf us
[J g−1 ]
Tcryst
[○C]
∆Hcryst
[J g−1 ]
33
56
181
28
105
–
33
33
32
32
34
34
32
53
57
55
57
57
57
57
57
17
16
17
14
15
16
15
16
9
15
12
11
11
15
15
11
12
15
15
14
15
17
15
14
Table 4.8: Melting and crystallization parameters of native dispersions. Values for
the bulk and trimyristin dispersion. *values are taken from literature [10]
+
Emulsifier composition of literature dispersion: 1.6 % S100, 0.4 % sodium
glycocholate.
The experimentally determined crystallization temperatures range from 10–15 ○C. Dispersions with a high co-stabilizer concentration of CPY or D2TAB crystallize both at
the same temperature as the respective dispersion with a low co-stabilizer concentration
(15 °C for CPY and 11 °C for D2TAB respectively) whereas dispersions with CTAB as
co-stabilizer display different crystallization temperatures for a high and low co-stabilizer
concentration which is 12 °C for low concentrations and 15 °C for high concentrations.
DTAB is only available at small co-stabilizer concentrations and crystallizes at 11 °C. For
low co-stabilizer concentrations a decrease of the melting temperature with decreasing
chain length of the co-stabilizer is observed.
The enthalpy values for the crystallization ∆Hcryst range between 14–17 J g−1 and are
comparable to the values of the enthalpy of fusion ∆Hf us for the melting curves, suggesting that crystallization and melting may occur in the same polymorphs. [10]
The crystallization temperature reported in the literature for the β-phase of bulk trimyristin is 28 ○C and for dispersed trimyristin particles 9 ○C respectively (Tab. 4.8). The
temperature shift to lower crystallization temperatures for the dispersed particles compared to the bulk is due to supercooling in the dispersed system. [10]
41
CHAPTER 4. RESULTS AND DISCUSSION
Comparing literature and experimental data for trimyristin dispersions similar values
are found. A temperature shift is also observed in the dispersions studied indicating the
expected supercooling behaviour. The experimental crystallization temperatures and
enthalpy values were slightly higher than values for the trimyristin dispersion reported
in the literature. However this might be ascribed to the fact that different stabilizing
agents are used by Bunjes. [10]
A general observation for the melting and crystallization behaviour of the trimyristin
dispersions is that the choice of co-stabilizer influenced the crystalline composition of the
dispersions and the ratio between the α- and β-phase. The amount of α-phase seems to
decrease with decreasing chain length of the co-stabilizer. This effect seems to be much
more pronounced at higher concentrations.
In conclusion, it can be stated that it was possible to produce dispersions of cationically modified trimyristin nanoparticles both by ultrasonication and high pressure melt
homogenisation. The particle sizes are around 220 nm for dispersions obtained by ultrasonication and 110-140 nm for dispersions obtained by high pressure melt homogenisation. Characterization of the particles by ζ-potential measurements, SAXS, WAXS and
DSC suggests that the particles crystallize predominantly in their β modification and exhibit self assembly tendency (stack formation). Cryo-electron microscopy pictures would
provide further insights on the particle shape and could also confirm stack formation.
4.3 DNA complexes of cationic modified triglyceride
nanosuspensions
The second part of the project was devoted to the preparation of DNA-trimyristin nanoparticle complexes. The approach chosen in this work is based on previous unpublished
work of A.Illing. [28] It can be assumed that the negatively charged DNA strongly interacts with the positively charged surface of triglyceride nanoparticles upon mixing of
DNA solution with the diluted dispersion of cationic modified trimyristin nanoparticles.
The formation of stack like complexes with alternating negative charged DNA and positively charged trimyristin particles was therefore expected.
The preparation strategy was to prepare mixtures with a varying DNA content from
low to high concentrations to increase the negative charge in the mixture and to study
the influence of the DNA concentration on the DNA-trimyristin nanoparticle mixtures.
The DNA concentration can be also be expressed as the charge ratio (CR) between the
42
CHAPTER 4. RESULTS AND DISCUSSION
positive charge of the cationic surfactant (DDAB) and the negative charge of the anionic
DNA, for example, CR 1 corresponds to a low DNA concentration where the ratio of
DNA is equal to the cationic surfactant whereas CR 0.3 corresponds to a high DNA
concentration where the ratio of DNA is abundant. The complex size was expected to
be influenced by the charge ratio.
4.3.1 Preparation
Samples with 5 different DNA-trimyristin nanoparticle ratios (CR 0.3, 0.5, 0.7, 1,2) were
prepared for all stable trimyristin dispersions prepared in the first part of this work.
Agglomeration was observed for all DNA-trimyristin nanoparticle mixtures prepared.
For mixtures with a high DNA concentration (CR 0.3, 0.5, 0.7) gel formation was observed in some cases. For low DNA concentrations (CR 1 and 2) the samples stayed
liquid. The strength of agglomeration was influenced by the co-stabilizer concentration
of the trimyristin dispersions as well. A higher co-stabilizer concentration (1.2 %) resulted in a stronger agglomeration as compared to dispersions with a low co-stabilizer
concentration of 0.4 %. Furthermore, for mixtures with a high co-stabilizer concentration
of D2TAB, a phase separation with sedimentation of microparticles can be observed.
The agglomerations made PCS measurement difficult as measurements of samples with
a particle sizes above 300 nm and a polydispersity index above 2.5 which indicates polydisperse samples become inaccurate. Table 4.9 summarizes the measured PCS values.
Nevertheless, the results suggest a decreasing particle size with decreasing DNA concentration. For very low DNA contents the size of DNA-trimyristin nanoparticle-complex
becomes similar to the particles size of the native dispersion. It was also observed that
the agglomeration tendency increases with the age of the trimyristin dispersions.
4.3.2 SAXS measurements
To obtain further information on the samples, they were also studied by SAXS. A high
and a low DNA concentration (CR 0.3 and CR 1, respectively) were chosen from previous
experiments and samples using the seven stable trimyristin dispersion with a high and
low co-stabilizer concentration were prepared at both DNA concentrations. The dispersions studied exhibit unusual SAXS patterns for both charge ratios differing significantly
from the patterns observed for the native dispersions (Fig. 4.5).
43
CHAPTER 4. RESULTS AND DISCUSSION
0 0 1 B ra g g p e a k
3
4
%
0 .4
0 .4
0 .4
1 .2
1 .2
1 .2
B
A B
B
A B
D N A
In te n s itä t
2
c o s ta b .
C P Y
C T A
D 2 T
C P Y
C T A
D 2 T
p u re
s = 0 .1 7
0 ,0 1
0 ,1
1
-1
s [n m
]
(a)
c o s ta b .
0 0 1 B ra g g p e a k
C P Y
D 2 T
D T A
C P Y
C T A
D 2 T
p u re
1
2
3
2
In te n s ity
1
1
A B
B
B
A B
D N A
%
0 .4
0 .4
0 .4
1 .2
1 .2
1 .2
3
2
3
s = 0 .1 7
0 ,0 1
0 ,1
s [n m
1
-1
]
(b)
Figure 4.5:
SAXS measurements of DNA loaded trimyristin dispersions for a) low DNA concentration CR 1 b)
high DNA concentration CR 0.3. The curves were shifted vertically for a better visualization; The
numbers mark suggestions of the order of the corresponding interference maximum due to stack
formulation.
44
CHAPTER 4. RESULTS AND DISCUSSION
CR
CPY, 0.4 %
d [nm]
CTAB, 0.4 %
d [nm]
D2TAB, 0.4 %
d [nm]
DTAB, 0.4 %
d [nm]
CPY, 1.2 %
d [nm]
CTAB, 1.2 %
d [nm]
0.3
0.5
0.7
1
2
0
6209.0
6291.0
5607.0
3036.0
1168.0
141.3
9351.0
4930.0
1017.0
> 500
> 500
137.1
30985.0
16676.0
14500.0
1210.0
448.0
147.5
2087.0
3029.0
4477.0
5624.0
> 500
312.0
6960.0
4343.0
3244.0
> 500
177.2
161.0
> 500
9233.0
4746.0
5309.0
323.1
166.7
Table 4.9: Summary of PCS values for DNA-trimyristin-mixtures. Values above ≥300 nm
with a polydispersity index above 2.5 which indicates polydisperse samples
have a limited significance. d is the z-avarage particle diameter.
All samples exhibit the 001 Bragg peak which is still visible at the same position (s ≈ 2, 8)
as for the native disersions. It was found at the same position independent of the charge
ratio, the composition and concentration of the co-stabilizer (Tab. 4.10). This can be
explained as the 001 Bragg reflection describes the length of the unit cell of trimyristin
and is influenced by the particle thickness, which is not influenced by the addition of
DNA.
Apart from the 001 Bragg reflection further interferences can be observed. The SAXS
patters are similar for low and high DNA concentrations but an influence of the costabilizer concentrations can be observed as dispersions with a low co-stabilizer concentration of 4 % display different interferences than dispersions with a high co-stabilizer
concentration of 1.2 %.
For dispersions with a low DNA concentration (CR 1) and a low co-stabilizer concentration (4 %) similar interferences as for the native dispersions are observed. In the native
state these were attributed to stack formation. Up to four orders can be observed and
they may be assigned to the second, third and forth order (Fig. 4.5(a)). Furthermore
a local minimum at low s-values (s ≈ 0.05) is observed. The observed interferences
can be attributed to the stack related interferences known from the native dispersions.
They indicate that some native dispersion remain beside the association with DNA. The
local minimum suggests the presence of another other underlying structure which can
be attributed to the DNA-trimyristin nanoparticle complexes. In addition it could be
interpreted as an absence of stack formation in the DNA-trimyristin complexes.
For dispersions with a low DNA concentration (CR 1) and a high co-stabilizer concentration of 1.2 %, instead of stack related interferences a single sharp peak at d ≈ 5.88 nm is
45
CHAPTER 4. RESULTS AND DISCUSSION
sample
CR 0.3
s001
[nm−1 ]
s(1st order)
[nm−1 ]
s(2st order)
[nm−1 ]
s(3st order)
[nm−1 ]
CPY, 0.4 %
CTAB, 0.4 %
D2TAB, 0.4 %
DTAB, 0.4 %
CPY, 1.2 %
CTAB, 1.2 %
D2TAB, 1.2 %
0.2746
0.2805
0,2850
0.2746
0.2850
0.2868
0.0498
0.0854
0.0993
-
0.0943
0.138
0.156
-
0.179
0.191
0.199
-
sample
CR 1
CPY, 0.4 %
CTAB, 0.4 %
D2TAB, 0.4 %
CPY, 1.2 %
CTAB, 1.2 %
D2TAB, 1.2 %
s001
[nm−1 ]
0.2820
0.2803
0.2803
0.2767
0.2840
0.2767
s(2st order)
[nm−1 ]
0.0994
0.0994
0.988
-
s(3st order)
[nm−1 ]
0.141
0.150
0.150
-
s(4st order)
[nm−1 ]
0.188
0.187
0.197
-
Table 4.10: Positions of the 001 Bragg reflection for dispersions of CR 0,3 and CR 1
respectively. In addition, s-values for the stack related interferences are
listed which are observed for low co-stabilizer concentration (0.4 %) only.
46
CHAPTER 4. RESULTS AND DISCUSSION
observed. Moreover, a local minimum at s ≈ 0.05 as for low co-stabilizer concentrations
is also observed for the co-stabilizers CPY and CTAB. A possibility for the interpretation of the sharp peak would be to assign the peak to free DNA in the solution but
comparison with SAXS-data of pure DNA solution showed that the peak for free DNA
should be found at much smaller s-values (Fig. 4.5(a)).
For dispersions with a high DNA concentration (Cr 0.3) similar patterns as for low
DNA concentrations are observed. A low co-stabilizer concentration (4 %) displays also
features that could be attributed to stack related interferences. They are less distinct
and up to three orders can be oberserved. For high co-stabilizer concentrations a similar
sharp single peak is observed as well at a slightly lower d-value (d ≈ 6, 25 nm).The peak
was also compared to SAXS data of pure DNA solution but was also not identified as
free DNA.
A possible explanation for the scattering pattern was found by comparison with a system of polymer clay nanocomposites which bears a certain resemblance to the presented
trimyristin dispersions. The described clay particle system contains of infinitely thin
disks carrying a point quadrupole, which results from the electric double layer around
the clay platelets. Simulations to reproduce experimental scattering data have been
carried out and compared to experimental data. The best matching simulations indicate gel like structures with particles arranged either perpendicular or parallel to each
other. [53,54] The simulated patterns show similarities to the recorded trimyristin-DNAcomplex SAXS patterns allowing a possible interpretation for the observed patterns
which indicate the formation of gel-like structures rather than the formation of stacks.
A reason for this behaviour might be the absence of Poloxamer 188 stabilizer which was
present in the previous study where DNA-tripalmitin stacks were observed. [28]
4.3.3 DSC measurements
Previous work has shown that the melting behaviour of DNA-tripalmitin nanoparticle
complexes is influenced by addition of DNA and it was expected to observe a similar
effect for the studied trimyristin-DNA-complexes.
Mixtures with a low DNA concentration (CR 1) and the native dispersions with a low
(0.4 %) and a high co-stabilizer concentration (1.2 %) were prepared and measured using
the Perkin Elmer DSC (Fig. 4.6). The results were compared to those obtained for native
dispersions (Fig. 4.4).
47
CHAPTER 4. RESULTS AND DISCUSSION
c o s ta b
2 0
- 5 8 ° C
B
A B
B
B
A B
%
0 .4
0 .4
0 .4
0 .4
1 .2
1 .2
1 .2
0
h e a tin g
c o o lin g
e n d o th e rm
N o r m a liz e d h e a tflo w [m W
/ g ]
5 0 ° C
C P Y
C T A
D 2 T
D T A
C P Y
C T A
D 2 T
-2 0
1 8 ° C -2 9 ° C
-4 0
2 0
4 0
6 0
T [° C ]
Figure 4.6: Power compensated DSC measurements for low DNA concentrations (CR 1)
(10 ○C/min). The upper curves show the heating cycle and the lower curves
show the cooling cycle.
For DNA loaded dispersions only one broad melting regime instead of two as for the
native dispersions was observed. The melting regime of the DNA loaded dispersions
was smaller compared to the native dispersions and no multiple melting events could be
distinguished within the broad melting curve. The average melting temperatures for the
dispersions with a low co-stabilizer concentration of 0.4 % were between 54 ○C and 55 ○C.
For the dispersions with 1.2 % costabiliser concentration the melting temperatures were
found to be between 55 ○C and 56 ○C. These temperatures are similar to the melting
temperatures of the native dispersions.
The melting enthalpy for DNA loaded dispersion ranges between 3–5 J g−1 . The melting enthalpy for dispersions with a high co-stabilizer concentration is the same for
all co-stabilizers (3 J g−1 ) whereas the enthalpy for a low co-stabilizer concentration is
slightly higher (3–5 J g−1 ) and increases with decreasing chain length of the co-stabilizer
(Tab. 4.11). Comparison with the enthalpy for the native dispersions which is between
14–17 J g−1 showed a much lower enthalpy for the DNA loaded dispersions and indicates
that addition of DNA influences the melting behaviour (Tab. 4.8).
The crystallization temperatures display a lower super cooling behaviour than the native
dispersions with higher crystallization temperatures between 22–28 ○C. A temperature
difference of 10 ○C between the native dispersions and the DNA containing samples was
48
CHAPTER 4. RESULTS AND DISCUSSION
sample
costab.
CPY
CTAB
D2TAB
DTAB
CPY
CTAB
D2TAB
%
0.4
0.4
0.4
0.4
1.2
1.2
1.2
%
%
%
%
%
%
%
Tm
[○C]
∆Hf us
[J g−1 ]
Tcryst
[○C]
55
54
54
54
57
57
56
3
4
4
5
3
3
3
22
22
23
25
24
28
28
∆Hcryst
[J g−1 ]
Table 4.11: Results for DSC measurements with the Perkin Elmer DSC of DNAtriglyceride nanoparticle complexes at CR 1.
observed. Comparison of the experimental crystallization temperatures for DNA loaded
dispersions with the crystallization temperature of the bulk (T = 28 ○C) known from literature displays a similar temperature range. Furthermore it was found that dispersions
with a co-stabilizer concentration of 1.2 % CTAB and 1.2 % D2TAB even crystallize at
the same temperature as the bulk (Tab. 4.11). For low co-stabilizer concentrations of
0.4% an increase of the crytsallization temperature with decreasing chain length of the
co-stabilizer is observed.
The observations on the melting and crystallization behaviour indicate that the agglomeration is also reflected in the thermal behaviour and the concentration of costabilizer influences the melting and crystallization behaviour. By agglomeration the
DNA-trimyristin mixtures seem to display a bulk like behaviour as the melting regime
is much narrower then for the native dispersions and the crystallization temperatures
are similar to the bulk crystallization temperature. In addition, for a low co-stabilizer
concentration an increase of the melting enthalpy and crystallization temperature with
decreasing chain length of the co-stabilizer, respectively is observed. This cannot be
observed for dispersions with a high co-stabilizer concentration of 1.2 %.
To study the influence of DNA concentration on the melting and crystallization behaviour different charge ratios for the same dispersion were prepared and their µ-DSC
measurements were compared to each other (Fig. 4.7). µ-DSC measurements were chosen
as they allow a higher resolution of the melting process.
The melting curves (Fig. 4.7(a)) showed the transformation of the discrete multiple
49
CHAPTER 4. RESULTS AND DISCUSSION
a-phase
b-phase
(a)
6
5
C R
0 ,3
C R
1
2
C R 5
n a tiv e
N o r m a liz e d h e a tflo w [m W ]
C R
4
3
2
1
0
1 0
2 0
3 0
4 0
T [° C ]
(b)
Figure 4.7:
µ-DSC measurement of CPY 0.4 % with different charge ratios a) melting curves b) cooling curves.
Curves are shifted for better overview.
50
CHAPTER 4. RESULTS AND DISCUSSION
melting events which were observed for the native dispersion transformed into a sharp
distinct melting peak at 55.7 ○C with increasing DNA concentration. For low DNA concentrations (CR 1, 2, 5) two major melting events as observed for the native dispersions
which are assigned to the melting of the α- and the β-phase were observed. The melting
event for the α-phase disappeared for a high DNA concentration (CR 0.3). Furthermore,
for a high DNA concentration (CR 0.3) a single sharp melting peak for the β-phase is
observed instead of multiple melting events. The melting temperature for a high DNA
concentration (CR 0.3) was observed to be the same as for bulk trimyristin.
The enthalpy displays a change in the melting behaviour as well as the enthalpy decreases
from 5 J g−1 for low DNA concentrations (CR 1, 2, 5) to 4 J g−1 for a high DNA concentration (CR 0.3). Comparing the enthalpy of DNA loaded dispersions to the enthalpy
of bulk trimyristin and the enthalpy of trimyristin dispersions a similarity between the
enthalpy of the dispersed trimyristin and the enthalpy of DNA loaded dispersions is
observed.
Tripalmitin dispersions in previous studies displayed a similar effect. [28] The disappearance of the α-phase and transformation of multiple discrete melting events into a sharp
melting peak with increasing DNA concentration were observed. A possible explanation is the increasing agglomeration of the nanoparticles with increasing DNA content.
The agglomeration of the nanoparticles seems to display a similar behaviour as bulk
trimyristin as the melting temperatures of the agglomerates were similar to the melting temperature of bulk trimyristin. Furthermore, the development of the single sharp
melting peak, which is ascribed to the bulk melting temperature, demonstrates the transformation of the dispersed β-phase into an agglomerated bulk-like phase. The amount
of bulk-like phase increased with increasing agglomeration.
Considering the enthalpy of fusion it was expected that the agglomerates should also
display similar values to the enthalpy of fusion found for bulk trimyristin. This was
not the case instead the enthalpy of the DNA loaded dispersions corresponds to the
enthalpy of the native trimyristin dispersions. This behaviour has not been observed
before and the enthalpy values contradict the melting behaviour. An explanation might
be that despite the agglomeration of the nanoparticles the surface of the agglomerated
nanoparticles stays large and does not decrease. Therefore, the amount of energy which
is needed to melt the agglomerates stays low.
The crystallization curves (Fig. 4.7(b)) are similar except for a very high DNA concentration (CR 0.3) (Tab. 4.12) as the melting temperature shifted from 14 ○C for low
51
CHAPTER 4. RESULTS AND DISCUSSION
CR 0.3
CR 1
CR 2
CR 5
native
Tm
∆Hf us
Tcryst
[○C]
56
56
57
57
57
[J g−1 ]
4.229
5.220
5.059
5.142
5.059
[○C]
38
14
14
15
14
∆Hcryst
[J g−1 ]
3.615
4.622
4.239
4.525
4.239
Table 4.12: Melting and crystallisation parameters for CPY, 0.4 % at different charge
ratios (0.3, 1, 2, 5).
DNA concentrations to 38 ○C. For low DNA concentrations CR 1 and CR 2 a small
second peak is observed which disappears for the even lower DNA concentration CR 5.
The enthalpy values of the small peak are similar to values of bulk samples and indicate a presence of the bulk-phase which disappears with decreasing DNA concentration.
As the enthalpy values stayed similar for all charge ratios it can be assumed that the
crystallization peak observed in all cases is ascribed to the crystallization of trimyristin
(Tab. 4.12).
In conclusion, it was attempted to prepare trimyristin-DNA-complexes. Characterization of the particles by SAXS suggests a formation of such complexes. But instead
of the expected stack formation of the particles a different structure has formed. A
possibility might be a gel-like structure instead of the formation of stacks. Despite the
formation of trimyristin-DNA-complexes they were not stable as an agglomeration of
the nanoparticles takes place and the properties of the dispersed nanoparticles are lost.
Characterization of the particles by DSC indicates that the agglomerates of nanoparticles
and DNA behave like bulk trimyristin. The contradictory enthalpy values suggest that
the agglomerates exhibit a large surface.
52
5 Conclusion and future prospects
In this thesis the preparation and characterization of trimyristin nanoparticles was studied.
In the first part of the project stable dispersions by HPMH and ultrasonication without
the additional stabilizer Poloxamer 188 used by Illing [28] were obtained. Four series
with four different co-stabilizers CPY, CTAB, D2TAB and DTAB were prepared. The
particle size was around 220 nm for ultrasonication and 110-140 nm for high pressure
melt homogenisation. They exhibited a ζ -potential between 40 mV and 80 mV. Characterization of the particles by SAXS and WAXS suggested that the particles crystallize
in the stable β-modification. Stack related interferences with inter-particle spacings of
50 nm for a low co-stabilizer concentration of 0.4 % and 40 nm for a costabilizer concentration of 1.2 % were obtained. The d-value of the 001 Bragg reflection was found to
be d ≈ 3, 62. DSC measurements showed multiple discrete melting events which can be
explained by a particle size effect known from literature. The results of the characterization suggest an influence of the co-stabilizer on the particle size and the crystalline
composition.
In the second part of the project the preparation of DNA-trimyristin nanoparticle complexes was studied. It can be concluded that it was not possible to produce stable complexes as strong aggregation was observed. Characterization of the complexes by SAXS
showed only weak indication for stack formation. The data suggested however the formation of a gel like structure. Previous work suggests that additional stabilizer Poloxamer
188 is required to allow a stack formation and subsequently stable complexes. [28] The
DSC heating curves exhibit a transformation from multiple distinct melting events into
a single sharp melting peak with increasing DNA concentration. The enthalpy values are
contradictory to the melting temperatures and suggest that the agglomerates still display
a large surface as the native dispersions instead of a decreased surface of bulk trimyristin.
The results of the characterization suggest an influence of the co-stabilizer concentration
on the melting and crystallization behaviour as well on the agglomerates.
53
CHAPTER 5. CONCLUSION AND FUTURE PROSPECTS
In general Cryo-electron microscopy pictures could provide further insights on the native
dispersions and the prepared DNA complexes. They could also confirm some conclusions
made in this work for example about the particle shape of the pure dispersion and their
self assembly behaviour or give further insights what happens when mixing DNA solution
and trimyristin dispersions.
Furthermore, a study on the influence of salt on stability of DNA-triglyceride complexes
could provide interesting results. Results reported in the literature suggest a better stability through additional salt. [55,56] The influence of the chain length of the co-stabilizing
agent on the stability of the pure dispersions and subsequently the resulting complexes
could be studied as well. In both cases the preparation by ultrasonication might be
interesting.
The usage of shorter DNA or exchange of DNA with short electrolyte chains could be
employed to study the influence of the chain length on the stability of DNA-triglyceride
complexes.
54
Bibliography
[1] A. Wretlind, J. Parent. Enter. Nutr. 1981, 5, 230–235.
[2] H. Bunjes, J. Pharm. Pharmacol. 2010, 62, 1637–1645.
[3] K. Westesen, Colloid Polym. Sci. 2000, 278, 608–618.
[4] B. Siekmann, K. Westesen, Pharmazie in unserer Zeit 1992, 21, 128–129.
[5] W. Mehnert, K. Mäder, Adv. Drug Delivery Rev. 2001, 47, 165–196.
[6] H. Bunjes, Curr. Opin. Colloid Interface Sci. 2011, 16, 405–411.
[7] K. Westesen, H. Bunjes, Int. J. Pharm. 1995, 115, 129–131.
[8] H. Bunjes, M. H. J. Koch, J. Controlled Release 2005, 107, 229–243.
[9] P. Lawler, P. Dimik in Food lipids (Eds.: C. C. Akoh, D. B. Min), M. Dekker, New
York, 2002, pp. 293–318.
[10] H. Bunjes, K. Westesen, M. H. J. Koch, Int. J. Pharm. 1996, 129, 159–173.
[11] H. Bunjes, M. J. Koch, K. Westesen in Molecular Organisation on Interfaces, Vol.
121 (Ed.: G. Lagaly), Springer Berlin Heidelberg, 2002, pp. 7–10.
[12] H. Bunjes, M. Drechsler, M. H. J. Koch, K. Westesen, Pharmaceutical Research
2001, 18, 287–293.
[13] A. van Langevelde, R. Peschar, H. Schenk, Acta Crystallogr. Sect. B: Struct. Sci.
2001, 57, 372–377.
[14] H. Bunjes, M. H. J. Koch, K. Westesen, Langmuir 2000, 16, 5234–5241.
[15] K. M. Rosenblatt, H. Bunjes, Mol. Pharmaceutics 2008, 6, 105–120.
[16] H. Bunjes, F. Steiniger, W. Richter, Langmuir 2007, 23, 4005–4011.
[17] K. Westesen, B. Siekmann, Int. J. Pharm. 1997, 151, 35–45.
55
Bibliography
[18] T. Unruh, H. Bunjes, K. Westesen, M. H. J. Koch, J. Phys. Chem. B 1999, 103,
10373–10377.
[19] T. Unruh, H. Bunjes, K. Westesen, M. H. J. Koch, Colloid. Polym. Sci. 2001, 279,
398–403.
[20] R. H. Müller, S. A. Runge, V. Ravelli, A. F. Thünemann, W. Mehnert, E. B. Souto,
Eur. J. Pharm. Biopharm. 2008, 68, 535–544.
[21] K. Tabatt, M. Sameti, C. Olbrich, R. H. Müller, C.-M. Lehr, Eur. J. Pharm. Biopharm. 2004.
[22] M. Garcia-Fuentes, D. Torres, M. J. Alonso, Int. J. Pharm. 2005, 296, 122–132.
[23] A. Illing, T. Unruh, Int. J. Pharm. 2004, 284, 123–131.
[24] H. Bunjes, M. H. J. Koch, K. Westesen, J. Pharm. Sci. 2003, 92, 1509–1520.
[25] H. Bunjes, T. Unruh, Adv. Drug Delivery Rev. 2007, 59, 379–402.
[26] A. Illing, T. Unruh, M. Koch, Pharm. Res. 2004, 21, 592–597.
[27] T. Unruh, K. Westesen, P. Bösecke, P. Lindner, M. H. J. Koch, Langmuir 2002,
18, 1796–1800.
[28] Illing, Andreas, unpublished, 2004.
[29] S.-D. Clas, C. R. Dalton, B. C. Hancock, Pharm. Sci. Technol. Today 1999, 2,
311–320.
[30] G. W. H. Höhne, H.-J. Flammersheim, W. Hemminger, Differential scanning calorimetry, Springer, Berlin [u.a.], 2nd ed., 2003.
[31] E. Gmelin, S. M. Sarge, Thermochim. Acta 2000, 347, 9–13.
[32] J. Als-Nielsen, D. McMorrow, Elements of modern X-ray physics, Wiley, Hoboken,
2nd ed., 2011.
[33] O. Glatter, O. Kratky, Small angle x-ray scattering, Academic Press, London and
and New York, 1982.
[34] Brian R. Pauw, “The Impossible Project” - How to do a perfect SAXS measurement,
6.April, 2011.
[35] T. Unruh, J. Appl. Crystallogr. 2007, 40, 1008–1018.
56
Bibliography
[36] R. H. Müller, R. Schuhmann, K. Thode, Teilchengrössenmessung in der Laborpraxis:
Kurzes Lehrbuch mit Einführung in die Theorie: Photonenkorrelationsspektroskopie
(PCS) - Laserdiffraktometrie (LD) - Coulter-Messprinzip (CM) und Messbeispielen
aus dem Schwerpunktbereich der Koaleszenzuntersuchungen, Wiss. Verl.-Ges., Stuttgart, 1996.
[37] W. Tscharnuter in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd,
2006.
[38] N. d. Jaeger, H. Demeyere, R. Finsy, R. Sneyers, J. Vanderdeelen, P. van der Meeren,
M. van Laethem, Part. Part. Syst. Char. 1991, 8, 179–186.
[39] M. M. M. Schmidt, W. Schärtl, Einführung in die makromolekulare Chemie –
Physikalische Chemie der Polymeren: Skript zur Vorlesung, 23.04.08.
[40] F. Hinze, S. Ripperger, M. Stintz, Chem. Ing. Tech. 1999, 71, 338–347.
[41] M. Instruments, Zeta Potential in 30 Minutes, Malvern instruments technical report, 2008.
[42] P. W. Atkins, C. A. Trapp, M. Zillgitt, Physikalische Chemie, VCH, Weinheim
[u.a.], 4th ed., 2006.
[43] T. C. Huang, H. Toraya, T. N. Blanton, Y. Wu, J. Appl. Crystallogr. 1993, 26,
180–184.
[44] L. Fan, M. Degen, S. Bendle, N. Grupido, J. Ilavsky, J. Phys. Conf. Ser. 2010, 247,
012005.
[45] F. Zhang, J. Ilavsky, G. G. Long, J. P. G. Quintana, A. J. Allen, P. R. Jemian,
Metall. Mater. Trans. A 2010, 41, 1151–1158.
[46] C. A. Dreiss, K. S. Jack, A. P. Parker, J. Appl. Crystallogr. 2006, 39, 32–38.
[47] A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch, D. Hausermann, High
Pressure Res. 1996, 14, 235–248.
[48] fit2dcorr, https://sourceforge.net/projects/fit2dcorr/.
[49] K. Westesen, B. Siekmann, M. H. J. Koch, Int. J. Pharm. 1993, 93, 189–199.
[50] C. Loistl, MSc thesis, Fachhochschule München, München, 2006.
[51] S. Joseph, H. Bunjes, J. Pharm. Sci. 2012, 101, 2479–2489.
57
Bibliography
[52] M. Schmiele, S. Busch, H. Morhenn, T. Schindler, M. Westermann, F. Steiniger,
A. Radulescu, P. Lindner, R. Schweins, P. Boesecke, and Tobias Unruh, Structural
Characterization of the Phospholipid Stabilizer Layer at the Solid-Liquid Interface of
Dispersed Triglyceride Nanocrystals with Small Angle X-ray and Neutron Scattering,
submitted to Phys. Rev. E, .
[53] R. Fartaria, N. Javid, R. A. Pethrick, J. J. Liggat, J. Sefcik, M. B. Sweatman 2011,
7, 9157.
[54] M. Dijkstra, J. Hansen, P. Madden, Phys. Rev. Lett. 1995, 75, 2236–2239.
[55] J. J. McManus, J. O. Rädler, K. A. Dawson, J. Phys. Chem. B 2003, 107, 9869–
9875.
[56] M.-L. Ainalem, N. Kristen, S. E. Edler, Karen J., T. Nylander, Langmuir 2010,
26, 4965–4976.
58
Acknowledgements
I would like to thank Prof. Dr. Tobias Unruh for the opportunity to to carry out this
work in his research group and for the opportunity to take part in the ECIS 2012 in
Malmö. This half year has been quite educating and broadened my horizon.
I also would like to thank Professor Dr. Alexander Böker for the supervision of this
project and making an external master thesis in Erlangen possible.
I would like to thank my supervisor, Martin Schmiele for the with the sample preparation and the high pressure homogeniser. For helpful explanation on the theoretical
background, data interpretation and latex editing. Sharing the office and special humour
has been an interesting experience.
I would like to thank my other Colleges in the working group: Torben Schindler for
helpful discussions and ”supervising” my trips to Garching, Munich; Heidrun Brückner
and Christian Bär for help in the lab regarding equipment and chemicals, repairing
broken cuvettes and an introduction to fast decalcifying.
I gratefully acknowledge the beam time at the SAXS instrument granted by the FRM
II in Munich and for the support of Armin Kriele (Hard-Soft-Matter Lab @ FRM II).
I also gratefully acknowledge the measurement time at the Zetasizer at the Institute of
Particle Technology, University of Erlangen-Nürnberg.
People from the institute of crystallography supported this work with proving advice on
everyday stuff and a good atmosphere.
Furthermore I would like to thank Mark Bispinghoff and my father for proofreading
and correcting my thesis. Mark is also thanked for friendship, moral support and useful
advice on working with latex.
Last but not least I would like to thank my family and friends for their support and
understanding.
59
Appendix
A. Source code from Python script
Example calculation for a DNA experiment. The charge ratios 0.3, 0.5, 0.7, 1.0 and
2.0 are prepared with a diluted trimyristin dispersion (3 % trimyristin ) and costabiliser
CPY.
# molar masses in g/mol
Mh=1.0; Mna=23.0; Mc=12.0; Mo=16.0
Mn=14.0; Mp=31.0; Mbr=79.9; Mcl=35.45
# CPY (cationic, 1+), C21H38NCl
Mcpy=21*Mc+38*Mh+Mn+Mcl # 339.45 Sigma: 339.99242
# DNA (anionic, 2-)
# MdnaAT=(5+5)*Mc+(4+5)*Mh+(5+2)*Mn+(0+2)*Mo+2*(5*Mc+7*Mh+5*Mo+Mp)
# MdnaGC=(4+5)*Mc+(4+4)*Mh+(3+5)*Mn+(1+1)*Mo+2*(5*Mc+7*Mh+5*Mo+Mp)
# Mdna=(MdnaAT+MdnaGC)/2.0
# DNA sodium salt
MdnaNaAT=(5+5)*Mc+(4+5)*Mh+(5+2)*Mn+(0+2)*Mo+2*(5*Mc+7*Mh+5*Mo+Mp)+2*Mna
MdnaNaGC=(4+5)*Mc+(4+4)*Mh+(3+5)*Mn+(1+1)*Mo+2*(5*Mc+7*Mh+5*Mo+Mp)+2*Mna
MdnaNa=(MdnaNaAT+MdnaNaGC)/2.0 # 330.75 x 2
# masses for native dispersion
m_myr = 8
m_S100 = 1.92
m_cpy = 0.32
m_H2O = 69.76
m_ges = m_myr + m_S100 + m_cpy + m_PLX + m_H2O
# m_filled_vial is the amount of added native dispersion
# rows x cols = 1 x ( different DNA contents )
60
Appendix
m_filled_vial = array([0.3,0.3,0.3,0.3,0.3,0.3])
m_ges_vial = m_filled_vial
m_myr_vial = ( m_myr / m_ges ) * m_filled_vial
m_cpy_vial = ( m_cpy / m_ges ) * m_filled_vial
m_H2O_vial = ( m_H2O / m_ges ) * m_filled_vial
cols=shape(m_filled_vial)[0]
# compute amount of DNA to be added, x_DNA_vial, to match the right CR
# (+/-) charge ratio #
pm=array([0.3,0.5,0.7,1.0,2.0,inf])
x_DNA_vial = m_cpy_vial / ( 2 * pm ) * ( MdnaNa/Mcpy )
# update m_ges_ud_vial
m_ges_vial = m_ges_vial + x_DNA_vial
#
#
#
#
compute amount of H2O to be added,
x_H2O_vial in order to fulfill
the right weight fraction of trimyristin, kappa
kappa == m_myr_vial / ( m_ges_vial + x_H2O_vial )
# mass fraction of trimyristin #
kappa=0.03
x_H2O_vial = m_myr_vial / kappa - m_ges_vial
# Check for mmyr:mDNA ratio
print("mass ratio mmyr:mDNA")
print(m_myr_vial/x_DNA_vial)
print("")
# check for kappa
print("kappa")
print(m_myr_vial / ( m_ges_vial + x_H2O_vial ))
print("")
# Prepare parent DNA-solution with H2O.
# For most of the other solutions additional H2O, y_H2O_vial, must be added.
dummy = x_DNA_vial/x_H2O_vial
parent_sol_H2O = where ( dummy==dummy.max() )
iH=parent_sol_H2O[0]
61
Appendix
# mass fractions of DNA and H2O in the H2O-DNA parent solution
kappa_DNA_parent_sol_H2O=x_DNA_vial[iH]/(x_DNA_vial[iH]+x_H2O_vial[iH])
kappa_H2O_parent_sol_H2O=x_H2O_vial[iH]/(x_DNA_vial[iH]+x_H2O_vial[iH])
# mass of H2O-DNA parent solution that must be added in order to fulfill the right DNA ma
m_parent_sol_H2O=zeros(cols,double)
# mass of H2O added with the parent solution
m_H2O_parent_sol_H2O=zeros(cols,double)
# masses of H2O still to be added
y_H2O_vial=zeros(cols,double)
for i in range(0, cols):
m_parent_sol_H2O[i] = x_DNA_vial[i] / kappa_DNA_parent_sol_H2O
m_H2O_parent_sol_H2O[i] = m_parent_sol_H2O[i] * kappa_H2O_parent_sol_H2O
y_H2O_vial[i] = x_H2O_vial[i] - m_H2O_parent_sol_H2O[i]
# print results
62