Progeria Syndrome - Research In Allied Health
advertisement

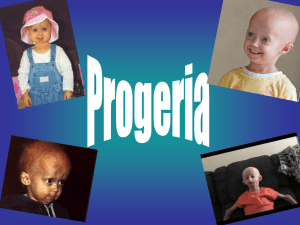
Progeria Syndrome Tamara Lansden East Tennessee State University Research in Allied Health Abstract Progeria syndrome is a rare and fatal genetic condition characterized by an appearance of rapid aging in children. Progeria syndrome is a disease that affects only one child in four million live births. These children born with the disease start out their lives looking and developing as a normal child does. However, within eighteen to twenty four months, the child begins to develop characteristics of progressed aging. The purpose of this study of research is to identify the different ages of children affected with progeria syndrome and how at that age, can cause stroke and heart disease. In this research I want to be able to detect an association with age and heart disease. I wish to be able to determine if manipulation of the Lamin A protein would provide some kind of depletion of the ageing process in already affected children. Introduction Progeria Syndrome is a rare, fatal genetic condition characterized by an appearance of rapid aging in children. Its name is derived from the Greek and means "prematurely old." While there are different forms of Progeria, the classic type is Hutchinson-Gilford Progeria Syndrome or HGPS. HGPS is caused by a mutation in the gene called LMNA (pronounced, lamin - a). The LMNA gene produces the Lamin A protein, which is the structural scaffolding that holds the nucleus of a cell together. Researchers now believe that the defective Lamin A protein makes the nucleus unstable. That cellular instability appears to lead to the process of premature aging in progeria. Although they are born looking healthy, children with progeria begin to display many characteristics of accelerated aging at around eighteen to twentyfour months of age. Signs of progeria include growth failure, loss of body fat and hair, aged-looking skin, stiffness of joints, hip dislocation, dry-scaly skin, limited range of motion, enlarged heart, and high blood pressure. These children have a remarkably similar appearance, despite differing ethnic background. Children with progeria usually develop and die from atherosclerosis, cardiovascular (heart) disease and stroke at an average age of thirteen years (with a range of about eight to twenty-one years). Although there is presently no treatment or cure for Progeria syndrome, the Progeria Research Foundation helped in the discovery of the gene that causes Progeria which is the first important step in finding a cure. I chose to study this disease because the disease is so rare that many people are not aware that it does exist. In my research I wish to be able to provide a better insight on the disease and be able to help individuals who have an interest in this particular study to understand that age is closely related to stroke and heart disease in children who are affected with Progeria Syndrome. Problem Statement In this research study, I want to look at heart disease and stroke as the dependent variables and I chose to identify age as the independent variable. In my research I want to be able to relate or prove that there is an association between the different ages of patients that have progeria syndrome and relate it to heart disease and stroke. The second part that I wanted to identify is how the treatment of this disease would prolong the livelihood of an affected child. Review of Literature The progeria syndrome is a rare genetic disorder, first reported in 1886 by Hutchinson and Guilford in England. The inheritance pattern, paternal age effect, and lack of consanguinity argue that it is due to a sporadic dominant mutation. Hutchinson-Guilford progeria syndrome (HGPS) is associated with several features of premature ageing--for example, growth retardation, characteristic facies, loss of hair, and subcutaneous fat, restricted joint mobility, prominent eyes, and severe premature atherosclerosis (Balin, p. 93). In 1886, Jonathan Hutchinson reported a case of a 3.5 year old boy who had the appearance of an old man. Later, Hastings Guilford reported a second case with similar features. The term, "progeria" was taken from the Greek word for old age, "geras", originally pro posed by Guilford in 1904 (Sarkar, 2001, p. 312). In 1972, DeBusk presented case reports of four patients and reviewed the world literature on HGPS. Although these patients develop premature atherosclerosis and die of cardiac or cerebral vascular disease between 7 and 27 years of age, many other features associated with pathological ageing are absent. DeBusk observed that patients “with progeria syndrome are typically considered normal infants at first.” The characteristic facies, posture, stiffness of joints, and bone, teeth, and skin changes become apparent during the second year of life (p. 697-700). Brown suggests that the estimated incidence of HGPS in the USA is one in eight million births, based on the number of cases. Brown also suggested that only one half of the affected patients were reported, thereby the estimated incidence of one in four million live births. Males are affected one and a half times more often than females (M : F = 1.5 : 1). Ninety seven per cent of affected patients are white. Several rare conditions exist in human beings that exhibit certain phenotypic characteristics associated with senescence. Often referred to as "segmental progeroid syndromes", the most important and widely studied condition was HGPS. The exact inheritance of HGPS is not known. In the past, it was thought to be an autosomal recessive (p. 293-294). DeBusk reviewed only three families where more than one member was affected, consanguinity was uncommon, and advanced paternal age was noted. These observations made an autosomal recessive inheritance very unlikely and favour a sporadic, dominant mutation (p.701). Since most cases are due to isolated mutations of a gamete, the familial occurrence is rare. Some cases may be due to germ line mutations, and therefore, parents of one affected child should be counseled that the risk of a subsequent affected child is one in 500 for each pregnancy" (Brown, p. 293). HGPS, a rare disease that speeds the aging process so much that children with the disorder die from diseases typically associated with advanced age, usually in their early teens. Since the 2003 discovery that a mutation in the LMNA gene causes progeria, scientists have developed several mouse models for the disorder, amassed a growing body of evidence regarding the disease's molecular and cellular basis, and devised experimental treatment strategies. Whether such treatments would be safe and effective for children with progeria remains to be determined, but scientists and advocates are excited by the swift pace of progress (Kuehn, 2006, p. 876). In particular, the fact that most patients with progeria die from severe and accelerated cardiovascular disease has led some researchers to believe that understanding this disorder could lead to a better understanding and associate typical cardiovascular disease in aging adults (p. 877). Age changes theory provided by the research of Bennett in 1995 suggested that age related changes occur throughout the whole lifespan, often involving deterioration of an anatomical/physiological nature. Many cellular functions appear to change with increasing age. Random molecular damage leading to accumulation of defects, for example, lipofuscin, causes age related changes in organs and systems that we recognise as ageing (p. 3-4). “With increasing age heart disease and many other organ function decline.” Similarly, ages from 40 to 70, show ageing brain, heart, lungs, bones, joints, muscles, and even immune systems all have impaired function. Therefore, progeria syndrome does support the hypothesis that at a DNA level in part, from an imbalance between DNA damage and repair. In some of these syndromes, malignant transformations occur due to severe chromosomal aberrations (p. 6-9). Arteriosclerosis, nephrosclerosis, myocardial fibrosis, and vascular calcifications are significant cardiovascular findings. Pesce studies of necropsy studies on patients with HGPS revealed gross abnormalities in skin, cardiovascular and cerebrovascular tissues, stroke, (p.162-163). Recently, magnetic resonance angiography demonstrated bilateral occlusion of internal carotid and vertebral artery origins. (p. 163). Pathological studies have demonstrated premature subintimal fibrosis in the blood vessels. As the child becomes more and more old they become more vulnerable to stroke, and heart disease as well many other contributing factors in the aging process. The most serious aspect of the disease, however, and the cause of death in >90% of cases, is rapid, progressive arterial occlusive disease, with death from myocardial infarction or stroke occurring at an average age of 13 years (range, 8– 21 years). Specifically, postmortem studies have identified profound loss of vascular smooth muscle cells (VSMC) in the medial layer of large arteries, such as the aorta and carotid arteries, with replacement by collagen and extracellular matrix. Superimposed on these medial changes have been generalized features of atherosclerosis with focal areas of calcification (Yang, p. 10291-94). By studying cells from patients with progeria and mice with progeria like disorders, scientists are beginning to sort out how the mutant LMNA gene causes damage at the cellular and molecular levels. According to Goldman, in the cells of progeria patients, the aberrant gene results in defective copies of a protein that is a precursor of the lamin A protein. The defective protein is called LA 50, or progerin. Unlike normal lamin A, this protein is unable to carry out its normal functions in the nucleus. In addition, progerin builds up in the nucleus from one cell generation to the next. This abnormal accumulation causes the nucleus to lose its structural integrity and induces a very lobulated shape (p. 8963-8964). Goldman and others hypothesize that beyond providing structural integrity to the nucleus, normal lamin A proteins provide a scaffolding within the nucleus that enables normal gene expression. In a pair of studies presented at the American Society for Cell Biology in San Francisco, Calif, in December, Goldman and colleagues provided evidence supporting this hypothesis. The results of one study suggested that progerin changes the organization of heterochromatic regions of the chromosome and that over many generations of cells heterochromatin was lost. In this study, the scientists used the inactive X chromosome (they used only cells from female progeria patients) as a "reporter" to track what was happening to heterochromatin in these cells. The team also has found that the location of the defective proteins in the cell may interfere with DNA replication during the cell cycle, which might explain the rapid accumulation of physiological changes associated with aging (p. 8967). In a study conducted by Vargo and colleagues, using the protein progerin, that contains a 50-aminoacid deletion within the carboxyterminal portion of the protein, suspected that protein farnesylation might be crucial for the targeting of progerin to the nuclear rim, and we hypothesized that blocking farnesylation with a farnesyltransferase inhibitor (FTI) would mislocalize progerin away from the nuclear rim and reduce the frequency of misshapen nuclei (3250). Method I found my participants for this research study (all 16-21 year old individuals of mixed ethnicity) at 2 area hospitals, Johnson City Medical Center and Welmont in Kingsport. I obtained consent (parental where needed) from the individuals after I had my study and instruments (survey questions) reviewed by the university IRB and completion of the CiTi training program. All conditions to protect and adhere to the participants confidentiality based upon HIPPA guidelines were observed as noted in the ETSU IRB webpage. The study did not include any invasive procedures and the IRB did not perceive any psychological manipulation or danger to study participants when they reviewed my methods and Scales of Measurement. To find an appropriate model, I replicated another study in my review of the literature. The data came from National Human Genome Research Institute to find out what factors can contribute to heart disease and stroke. HGPS is caused by an LMNA mutation that results in the synthesis of a mutant prelamin A, commonly called progerin, that contains a 50-amino-acid deletion within the carboxyterminal portion of the protein. The age group of the children analyzed was from 7-10 years. The survey followed the health history, lab results, and symptoms from 4 years to 16 years of age. There were 3 subgroups within the randomized sample, group 1 included children ages 4- 9 years that received manipulation on the protein farnesylation, group 2 included children ages 10-16 years that received manipulation on the protein farnesylation, and group 3 included children ages 4-16 years that received no manipulation of the gene (Varga, 2006). I administered a potent FTI, ABT-100 (17), in the drinking water (39 mg/kg body weight) to both groups 1and 2 both male and female and the 3 group received no potent. There was one child selected for each mother so there was no relation between the participants. The outcome variables included questions such as: how many times has your child suffered from a minor stroke? How many times have your child suffered from a minor heart attack? Following the American Association of Pediatrics (AAP) guidelines, we dichotomized the outcome variable as 0 versus < 1 symptom per year for children 4-9 years of age, and 0 to 2 versus >3 symptoms per year for children 10 years or older. The Spearman correlation between year and a lifespan of symptoms was >0.8 for each age group and the average amount of heart failure in a lifespan was not the same as in a typical year for all ages. The independent variable I chose to identify is age. The analysis of the cross-sectional sample used the x2 test and the t test to analyze the independent variables and heart disease. Any predictors that were significantly (P<.05) correlated with heart failure in the bivariate analyses were included in multivariate logistic regression models that examined the odds of suffering heart disease symptoms <1 symptom for the youngest and middle groups and the odds of suffering >3 symptoms a year over a lifespan at age 21. The predictor of interest was heart failure or heart disease as a teen or young adult, but models also controlled for race, gender, child age and birth order, and multiple affected siblings (Varga, 2006). In my study I found that, using a Likert scale response method, teens who suffered more than 1 symptom related to heart disease or stroke, predictably, suffered from at least 3 symptoms related to heart disease per year at the age of 21. I suspected that protein farnesylation might be crucial for the targeting of progerin, and hypothesized that blocking farnesylation with a farnesyltransferase inhibitor (FTI) would mislocalize progerin in individuals and slow down or alter profound loss of vascular smooth muscle cells (VSMC) in the medial layer of large arteries, such as the aorta and carotid arteries contributing factors associated with the aging process, and heart disease. Furthermore, I found that these high risk individuals that did not undergo manipulation of the gene (blocking farnesylation) in their teens developed heart attacks, and suffered from stoke related illnesses than individuals affected by the mutation or manipulation of the gene, at the same age. Discussion After completing my research I have been able to prove my theory that there is an association between age and heart disease of affected individuals; however, research has not proved that manipulation of the Lamin A protein may cause a treatment, reverse or even deplete the progeria syndrome. Through progression of future research we can one day come to this magnificent discovery. Conclusion In this research study, I wanted to look at heart disease and stroke as the dependent variables and I chose to identify age as the independent variable. In my research paper I wanted to be able to relate and I did prove that there is an association between the different ages of patients that have progeria syndrome that suffers from heart disease and stroke. The second part that I wanted to identify but was not able to conclude is how manipulation of a progerin gene may lead to the treatment of this disease and furthermore would prolong the livelihood of an affected child. I would like to one day further my studies of this particular topic and I think that others interested in this area would like to progress with research as it develops as well to one treating Progeria syndrome. Reference Balin AD, ed. “Contribution of in vitro skin fibroblast studies from individuals with genetic disease that predispose to accelerated ageing phenomena to our understanding of the ageing process”. New York: Raven Press, 1989: 93119. Bennett GCJ, Ebrahim S. “The essentials of health care in old age”. (2nd ed.) New York: Oxford University Press, 1995: 3-10. Brown, W.T., ed al. "Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome." Nature. Vol. 423 (15 May 2003): p. 293-296. DeBusk FL. “The Hutchinson- Gilford progeria syndrome.” Pediatric Journal. Vol. 90 (1972): p. 697-724. Goldman, Robert D, et al. “Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome.” Proceedings of the National Academy of Sciences of the United States of America. Vol. 101:24 (June 2004): p. 8963-8968 Kuehn, Bridget M. “Gene discovery speeds progeria research”. Journal of the American Medical Association. Vol. 295 No. 822 (February 2006): p. 876878. Pesce K, Rothe MJ. “The premature ageing syndromes.” Clin Dermatol. Vol. 14 (1996): p. 161-70. Sarkar, P K, and R A Shinton. "Hutchinson-Guilford progeria syndrome." Postgraduate Medical Journal. Vol. 77.907 (May 2001):p. 312. Vargo, Renee, et al. “Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson- Gilford progeria syndrome.” Proceedings of the National Academy of Sciences of the United States of America. Vol.103:9 (28 February 2006): p. 3250-3255. Retrieved from: http://www.pnas.org/cgi/content/full/103/9/3250. Journal URL: http://ejournals.ebsco.com/direct.asp?ArticleID=4DAA925D27CDFF6307AA Yang, Shao H, Martin O. Bergo, et al. “Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson–Gilford progeria syndrome mutation.” Proceedings of the National Academy of Sciences of the United States of America. Volume 102, Number 29 (July 2005): p. 10291-10296.