Clinical Practice Essay - University of Rochester Pulmonary
advertisement

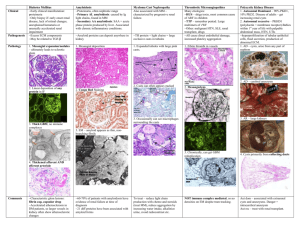
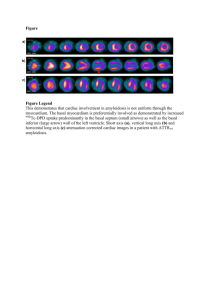
Nicholas Braus, MS2 School of Medicine and Dentistry University of Rochester 4.4.08 Clinical Practice Essay DL is a 56 year-old female who complained of shortness of breath on exertion over the last 6 months. She had been a smoker and therefore pulmonary function tests were done and showed a pattern typical of moderate restrictive lung disease (FEV1 of 58% predicted, FVC of 65% predicted). A chest radiograph showed normal lung fields. A CT scan was also performed, which showed diffuse groundglass infiltrates bilaterally. There was no adenopathy. A bronchoscopy and trans-bronchial biopsy was done that revealed normal airways, but thickened alveolar septae with amorphous eosinophilic deposition, positive for Congo red and Crystal violet staining, consistent with amyloidosis. Serum protein electrophoresis indicated a paraprotein in the near gamma region in a concentration of 1.6 g/dL exhibiting IgG Lambda reactivity. Urine electrophoresis showed free light chains. What further evaluation is needed and how should her care be managed? The Clinical Problem Amyloidosis describes a constellation of rare diseases that arise as a result of the deposition of a pathologic protinaceous substance in extracellular tissue. Tissues commonly involved include the kidneys (33%), heart (25%), peripheral and autonomic nerves (17%), gastrointestinal tract (8%), skin, joints, and blood vessels.1 The protean clinical features of amyloidosis, together with its biochemical and pathological heterogeneity, make its epidemiology challenging to define with precision. Classification of the disease is based on the biochemical profile of the precursor proteins that give rise to abnormal fibrillar protein deposits. Epidemiology & Nosology of Systemic Amyloidoses Of the systemic forms of the disease, so called “AL” or “Primary Amyloidosis” is the most common and arises from the deposition of amyloidogenic monoclonal immunoglobulin light chains. It has an age-adjusted incidence of 8.9 per million person-years and accounts for approximately 60% of all cases of amyloidosis.2 AL amyloidosis can occur in association with any form of monoclonal B cell dyscrasia, and it occurs in 12-15% of patients with multiple myeloma.3 Another systemic variant is “AA” or “Secondary Amyloidosis”, which derives from cleavage, misfolding and aggregation of the acute phase reactant serum amyloid A protein. It is a potential complication of any chronic inflammatory state and accounts for approximately 10% of cases.4 Familial Amyloidoses are a group of autosomal 1 dominant diseases in which amyloid deposition is derived from a mutant form of transthyretin (TTR), a transport protein for thyroxin and retinol-binding protein. Senile cardiac amyloidosis and familial amyloid cardiomyopathy are both associated with abnormal transthyretin. The incidence of familial transthyretin-associated amyloidosis is unknown, however it has been reported that the number of cases diagnosed in referral centers is approximately 10-20% that of AL amyloidosis.5 One analysis of the amyloidogenic transthyretin Ile 122 mutation found an allelic frequency of 0.020 in black Americans, indicating that 1.3 million black Americans carry the allele.6 Patients on long-term hemodialysis are at risk for an amyloidosis that derives from the deposition of circulating -2 microglobulin in subchondral bone and the synovia of joints and tendons. The prevalence of this type of amyloidosis has been observed in over 50% of patients at 165 months of dialysis treatment and approaches 100% in patients with over 20 years of treatment.7 Common manifestations of -2m amyloidosis include carpal tunnel syndrome and bone cysts. Finally, deposition of -amyloid (A) in brain tissue has been demonstrated to play a central role in the pathophysiology of Alzheimer’s Disease, which affects over 4.5 million Americans.8 -amyloid is a metabolic byproduct of “amyloid precursor protein” (APP), an integral membrane protein expressed in many tissues and concentrated in the synapses of neurons.9 Pathophysiology of Amyloid Deposition As noted above, the central pathophysiologic mechanism in systemic amyloidosis is formation and deposition of amyloid proteins in extracellular tissues. Despite its uniform appearance and tinctorial characteristics under electron, light, and polarizing microscopy, “amyloid” is not a chemically distinct entity. Rather, it describes a family of over 20 proteins that assume an insoluble -pleated fibrillar conformation under certain conditions. It is precisely this underlying biochemical heterogeneity of amyloid that gives rise to the disease’s strikingly heterogeneous clinical phenotypes. The molecular mechanism that conjugates precursor proteins into -fibrils is closely related to physiologic protein folding, in which newly synthesized peptide chains undergo a rapid sequence of conformational changes in the cytoplasm and become either native or misfolded proteins. The intrinsic 2 propensity for a protein to assume an amyloidogenic conformation is determined by the interaction of pro-fibrillar and anti-fibrillar characteristics. An abundance of residues favoring -pleated sheet conformation, poor stability of the protein’s native conformation, abundant hydrophobic side chains, and neutralization of charges on amino acid residues have all been identified as pro-fibrillar attributes of amyloidogenic proteins, while interruption of -sheets with residues like proline and the repulsion of amino acid residues with the same charge are characteristics that inhibit fribrillogeneis. 10 Beyond these intrinsic qualities, proteolytic remodeling and mutation have also been proposed as mechanisms of fibrillogenesis.11 Mutations contribute to amyloidogenicity by destabilizing critical regions of a protein’s structure and rendering it prone to fibrillar aggregation. For example, during the immune response lambda light chain variable domains of immunoglobulins mutate extensively and a small fraction of the final light chain proteins produced by this process are amyloidogenic. When such end products are produced extensively—as in the case of myeloma—they aggregate and deposit in tissue, and primary (AL) amyloidosis is the result. The role of proteolysis in the pathogenesis of amyloid deposition disorders is exemplified by the cleavage of the 753-residue amyloid precursor protein (APP) into fragments of 39 to 43 residues, which make up the amyloid plaques found in Alzheimer’s Disease. Additional non-fibrillar components of the molecular mechanism of amyloid production are the glycoprotein “serum amyloid P” and proteoglycans like haparan and and dermatan sulfate. These components are expressed in all amyloid deposits, regardless of the protein of origin or the tissue involved. Proteoglycans make up the carbohydrate component of amyloid, and it has been postulated that they act as a scaffolding to stabilize amyloid fibrils and facilitate aggregation. 12 Serum amyloid P (SAP) binds to a site common to all conformational varieties of amyloid, which has not yet been elucidated. SAP is considered important in the pathogenesis of amyloidosis because of it’s demonstrated ability to protect misfolded protein from degradation by proteases and resorption via normal cellular mechanisms of protein homeostasis. 13 The development of radio-labeled SAP 3 scintigraphy has led to advances in diagnosis and treatment by allowing non-invasive imaging and quantitative monitoring of disease progression.14 The pathological effects of amyloid deposition are mainly a result of the physical presence of excess non-soluble proteinaceous material in affected tissues. Normally, inappropriately aggregated or misfolded extracellular proteins are degraded by macrophages. In amyloidosis, however, normal molecular surveillance and proteolytic mechanisms fail, and serum amyloid proteins accumulate, aggregate, and are ultimately deposited in tissues. Accumulation of substantial protein deposits can disrupt tissue architecture, disrupt normal vessel wall dynamics, create space-occupying lesions, and impair organ function. Smaller deposits in specific tissues (e.g. glomeruli, cardiac conductive fibers, nerves) can also produce significant pathology. Although prognosis correlates well with overall amyloid burden, there is little evidence to support a correlation between severity of symptoms and the scale of deposits.15 Interestingly, histological evidence of inflammation is rare in amyloidosis. There is, however, some evidence from experiments in vitro that newly forming amyloid fibrils are cytotoxic and induce cell death by apoptosis and necrosis.16 This recent discovery is consistent with the reported clinical correlation between periods of active amyloid deposition and accelerated deterioration of tissues.17 Traditionally the natural history of amyloidosis has been understood as an unremitting and irreversible process of protein deposition with progressive functional decline of involved tissues. A population-based study of primary (AL) amyloidosis at Mayo Clinic reported a median survival time after diagnosis of 2.3 years.18 Contrary to the irreversible progression of the disease, however, serial SAP scintigraphic studies shown that amyloid deposits exist in a state of dynamic turnover. AL, AA, 2m, and TTR amyloid deposits have all been demonstrated to regress when the supply of precursor proteins is eliminated.19,20,21,22 This discovery is encouraging, although its implications for the treatment of amyloid deposition disorders is not yet clear. 4 Amyloidosis in the respiratory tract As first recorded by Virchow and Lesser in 1857, amyloidosis may affect any structure within the thorax including the lung parenchyma and respiratory tract.23 Pulmonary involvement in systemic disease is common and almost exclusively associated with primary (AL) amyloidosis. In fact, microscopic deposits of amyloid in the lungs are almost universally present at autopsy in patients with AL amyloidosis.24, 25 Clinically, however, this association is not always apparent as less than 10-20% of patients with AL amyloidosis present with significant respiratory symptoms. In addition, it has been demonstrated that in most cases amyloid-associated dyspnea is secondary to cardiac involvement.26 Since the advent of antimicrobial therapy, the prevalence of secondary (AA) pulmonary amyloidosis has declined significantly. Today the respiratory diseases underlying secondary pulmonary amyloidosis are bronchiectasis, tuberculosis, and Mediterranean fever.27 Pulmonary TTR-associated amyloidosis has been rarely reported and its clinical significance, if any, has yet to be established.28 Focal amyloidoses of the respiratory tract are almost exlusively of the AL type, and can be seen in the larynx, trachea, large bronchi, and the parenchymal tissue of the lung. Localized lesions, or “amyloidomas,” are almost never manifestations of systemic primary amyloidosis. Whereas amyloidogenic light chains in systemic AL amyloidosis clearly derive from circulating monoclonal light chains produced by plasma cell dyscrasias, studies of localized AL amyloidoses demonstrate that light chains in this type arise from very subtle focal lymphoplasmacytic proliferation confined to areas in proximity to amyloid deposits.29 Despite this observation, it is recommended that all patients with focal lesions be evaluated for signs of systemic disease.30 Amyloid deposition within the lung parenchyma, sometimes referred to as “alveolar septal amyloidosis,” stands out from other focal amyloidoses of the airways because of its two distinct presentations, each with separate epidemiological and pathophysiological profiles. Nodular amyloidosis in the lung parenchyma is usually an incidental finding and is not associated with systemic disease. Diffuse alveolar septal amyloidosis is usually a manifestation of systemic AL amyloidosis. 31 As 5 mentioned above, diffuse involvement of the lung parenchyma is considered the rule rather than the exception in systemic primary (AL) amyloidosis, making it by far the most prevalent form of pulmonary amyloidosis. Strategies and Evidence Diagnosis Diffuse alveolar septal amyloidosis usually presents with a history of progressive dyspnea and cough. Anatomical and functional evaluation should include plain radiographic and CT studies of the chest, bronchoscopy and pulmonary function tests. Plain films of the chest typically show diffuse reticulo-nodular infiltrates and occasionally hilar and mediastinal lymph nodes. CT scans show widespread interstitial involvement, occasionally with calcification.32 Pulmonary function tests may demonstrate a decline in lung volumes and diffusion capacity. The gold standard for diagnosis of amyloidosis is the apple-green birefringence seen when involved tissue is stained with Congo red dye and viewed under polarized light.33 Tissue specimens can be collected via small needle biopsy, though compared to open surgical resection this method entails significant sampling error.34 A reported risk of endo- and transbronchial biopsy is excessive bleeding due to increased fragility of involved blood vessles, reduced elasticity of involved parenchymal tissue, and very rarely an acquired clotting factor deficiency.35 When tissue biopsy is contraindicated, an effective alternative is abdominal fat pad aspiration cytology.36 Positive histology does not, however, provide information regarding the underlying protein type, systemic involvement, or the natural history of the disease. Identification of underlying systemic disease, including clonal plasma cell dyscrasias, must be sought via hematological and biochemical assays such as serum and urine protein electrophoresis. Due to the prevalence of monoclonal gammopathy of undertermined significance (MGUS), demonstration of monoclonal gammopathy and serum or urine light chains is not definitive evidence of AL amyloidosis and should not preclude immunohistochemical analysis to determine the protein of origin. In the absence of a plasma cell dyscrasia and 6 immunohistochemical evidence of AA protein, evidence of a transthyretin-associated amyloidosis should be sought using isoelectric focusing of the serum for abnormal TTR.37 Additional testing in the case of systemic disease includes tests for organ involvement such as liver enzyme assays, nerve conduction studies, and echocardiography. Finally, routine monitoring of the whole body load and distribution of amyloid deposits should be carried out using radiolabeled SAP scintigraphy. Intravenous radiolabeled SAP localizes to areas with high concentrations of amyloid-associated SAP, and facilitates the determination of the disease’s natural history and response to treatment in individual cases.38 Treatment Treatment options for alveolar septal AL amyloidosis are limited, and due to the paucity of randomized controlled trials treatment decisions are typically made on an empirical basis. Management is oriented around preservation of organ function and aggressive measures to reduce the rate of protein deposition. Treatment with steroids and irradiation has not been proven to influence the course of the disease, however in AL amyloidosis chemotherapy with combination melphalan and prednisone has been demonstrated to suppress the underlying dyscrasia and slow the rate of deposition in up to 30% of patients.39 Cytotoxic regimens have been reported to increase median survival time from 15 months in untreated patients to over 5 years in those receiving treatment.40 A recent study has also demonstrated the effectiveness of vincristine, doxorubicin, and dexamethasone (VAD) alone in high-risk patients with AL amyloidosis-related nephrotic syndrome.41 Autologous stem cell and bone marrow transplantation protocols have been developed with mixed success. A major limiting factor in these therapies is a relatively high treatment-related mortality rate, which is estimated to be between 10-20%.42 Areas of Uncertainty Fibrillogenesis stands out as an area of basic science research with great potential to contribute to the diagnosis and care of patients with amyloidoses, including Alzheimer’s Disease and perhaps even related proteinoses like prion diseases. A greater understanding the basic mechanisms determining the molecular stability of proteins and their propensity to assume pathological conformations has begun to 7 point medical research towards strategies for inhibiting the process. Research into these mechanisms has focused on the role of proteoglycans, chaperonins, and glycosaminoglycans (SAP) in stabilizing proteins in misfolded states.43 In addition, the pursuit of small molecule inhibitors of amyloid fibrillogenesis is very active in the development of new therapies for Alzheimer’s Disease.44 Conclusions and Recommendations Primary (AL) amylodiosis is a rare protein-deposition disorder caused by a monoclonal plasma cell dyscrasia. Clinically it follows a progressive and unremitting course with poor prognosis and limited treatment options. For patients with symptomatic organ dysfunction, such as the woman in the vignette, treatment should be guided by accurate characterization of the underlying proteinopathy with immunohistochemical staining, and the extent of her dyscrasia with peripheral blood smear and bone marrow biopsy. I would monitor her lung function and rigorously screen for extrapulmonary organ dysfunction, paying close attention to the kidneys, heart and liver. I would also routinely monitor her whole-body load and distribution of amyloid deposits with radiolabelled SAP scintigraphy. In one univariate analysis, bone marrow plasma cell percentage over 10%, circulating plasma cell percentage over 1%, and peripheral blood plasma cell count over 500,000/L (0.5/miroL), and prominent cardiac involvement each predicted for significantly worse overall survival. 45 Combination chemotherapy with melphalan and prednisone is recommended. Additional treatment modalities such as hematopoietic cell transplant and research protocols for combination chemotherapy and/or hematopoietic cell transplant may be considered. 1 Falk R., Comenzo R., and Skinner M. “The Systemic Amyloidoses.” NEJM 1997; 337:898-909. 2 Kyle R., Linos A., Beard C., et al. “Incidence and natural history of primary systemic amyloidosis in Olmstead County, MN, 1950 through 1989.” Blood 1992; 79:1817-22. 3 Bellotti V., Nuvolone M., et al. “The workings of the amyloid diseases.” Ann Med 2007; 39:200-207. 4 Gertz M., Kyle R., “Secondary systemic amyloidosis: response and survival in 64 patients.” Medicine (Baltimore) 1991; 70:246-56. 5 Falk, et al. 1997. 6 Jacobson D., Pastore R., et al. “Variant-sequence transthyretin (Ise 122) in late-onset cardiac amyloidosis in black Americans.” NEJM 1997; 336:466-473. 8 Charra B., Calemard E., Laurent G. “Chronic renal failure treatment duration and mode: their relevance to the late dialysis periarticular syndrome.” Blood Purif 1988; 6:117-124. 8 Hebert L., Scherr P., et al. “Alzheimer Disease in the US population: prevalence estimates using the 2000 census.” Arch Neurol 2003; 60:1119-1122. 9 Hardy J., Selkoe D. “The amyloid hypothesis of Alzheimer’s Disease: progress and problems on the road to therapeutics.” Science 2002; 297:353-356. 10 Bellotti, et al. 2007. 11 Merlini G., Bellotti, V. “Molecular Mechanisms of Amyloidosis.” NEJM 2003; 349(6):583-596. 12 Kisilevsky R., Ancsin J., Szarek W., Petanceska S. “Heparan sulfate as a therapeutic target in amyloidogenesis: prospects and possible complications.” Amyloid 2007; 14(1):21-32. 13 Tennent G., Lovat L., Pepys M. “Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci USA 1995; 92:4299-303. 14 Hawkins P., Lavender J., Pepys. “Evaluation of systemic amyloidosis by scintigraphy with 123!-labeled serum amyloid P component.” NEJM 1990; 323(8):508-13. 15 Pepys, M. “Pathogenesis, diagnosis and treatment of systemic amyloidosis.” Phil Trans R Soc Lond 2001; 356:203-211. 16 Gharibyan, A., Zamotin, V. “Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways.” J Molec Bio 2007; 365(5):13371349. 17 Pepys. 2001. 18 Utz J., Swenson S., Gerz M. “Pulmonary Amyloidosis: the Mayo Clinic experience from 1980 to 1993.” Ann Intern Med 1996; 124:407-413. 19 Hawkins P., Richardson S., et al. “Serum amyloid P component scintigraphy and turnover studies for diagnosis and quantitative monitoring of AA amyloidosis in juvenile rheumatoid arthritis.” Arthritis Rheum 1993; 36:842-851. 20 Hawkins P., Richardson S., et al. “Scintigraphic quantitation and serial monitoring of human visceral amyloid deposits provide evidence for turnover and regression.” Q J Med 1993; 86:365-374. 21 Rydh A., Suhr O., et al. “Serum amyloid P component scintigraphy in familial amyloid polyneuropathy: regression of visceral amyloid following liver transplantation.” Eur J Nucl Med; 25:709-713. 22 Tan S., Irish A., et al. “Long term effect of renal transplantation on dialysis-related amyloid deposits and symptomatology.” Kidney Int; 50:282-289. 23 Lesser A. “Ein fall von Enchondroma osteiodes mextum der lunge mit patieller amyloid entortung.” Virchows Arch (Pathol Anat) 1887; 69:404-8. 24 Utz, Swenson, and Gerz. 1996. 25 Smith R., Hutchins G., Moore G., Humphrey R. “Type and distribution of pulmonary parenchymal and vascular amyloid. Correlation with cardiac amyloidosis.” Am J Med 1979; 66:96-104. 26 Eshaghian S., Kaul S., Shah P. “Cardiac amyloidosis: new insights into diagnosis and management.” Rev Cardivasc Med 2007; 8(4):189-99. 27 Joss N., McLaughlin K., Simpson K., et al. “Presentation, survival, and prognostic markers in AA amyloidosis.” QJM 2000; 93:535-542. 28 Kunze, W. “Senile pulmonary amyloidosis.” Pathol Res Pract 1979; 264(4):413-22. 29 Glenner G. “Amyloid deposits and amyloidosis: the -fibrilloses. II. NEJM 1980; 302:1333-43. 7 9 Shah P., Gillmore J., Copley S. “The importance of complete screening for amyloid fibril type and systemic disease in patients with amyloidosis in the respiratory tract.” Sarcoidosis Vasc Diffuse Lung Dis 2002; 19:134-42. 31 Gillmore J., Hawkins P. “Amyloidosis and the respiratory tract.” Thorax 1999; 54:444451. 32 Graham C., Stearn E., et al. “High resolution CT appearance of diffuse alveolar septal amyloidosis.” Am J Roentgenol 1992; 158:265-7. 33 Gillmore and Hawkins, 1999. 34 Gillmore and Hawkins, 1999. 35 Lachmann H., Hawkins P. “Amyloidosis and the lung.” Chronic Respiratory Disease 2006; 3:203-214. 36 Dhingra S., Krishnani N., Kumari N., Pandey R. “Evaluation of abdominal fat pad aspiration cytology and grading for detection in systemic amyloidosis.” Acta Cytol 2007: 51(6):860-4. 37 Falk, et al. 1997. 38 Hawkins, Lavender, and Pepys. 1990. 39 Kyle R., Gertz M., et al. “A trial of three regimens for primary amyloidosis: colchicine alone melphalan and prednisone, and melphalan, prednisone, and colchicine.” NEJM 1997; 336(17):1202-1207. 40 Pepys, 2001. 41 Matsuda M., Gono T., et al. “Nephrotic syndrome due to primary systemic AL amyloidosis, successfully treated with VAD alone.” Intern Med 2008; 47(6):543-9. 42 Dimopoulos M. “Treatment of AL amyloidosis with high dose therpy and autologous stem cell transplantation.” Leukemia and Lymphoma 2006; 47(6):963-964. 43 Pepys, 2001. 44 Nitz M., Fenili D., et al. “Modulation of amyloid- aggregation and toxicity by inosose stereoisomers.” FEBS J 2008; 275(8):1663-74. 45 Pardanani A., Witzig A., et al. “Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis.” Blood 2003; 101(3):827-30. 30 10