Overlap Extension PCR Protocol (Fusion PCR)
advertisement
")
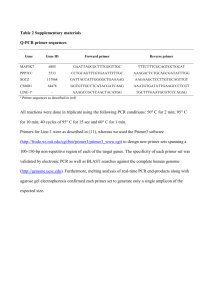
Overlap Extension PCR (a.k.a. Fusion PCR) References: Roche Expand Long Template Handout Sambrook & Russel. “Site Specific Mutagenesis by Overlap Extension” Molecular Cloning. CSH Protocols; 2006; doi:10.1101/pdb.prot3468 Purpose: To amplify fused pieces of DNA from two separate sources. This protcol is particularly handy for adding promoters or tags to a given gene sequence. The fused pieces can easily be cloned into a vector for amplification and sequencing or transformed directly into yeast. This protocol can also be modified to generate site specific mutations within a given gene by engineering the desired mutation into both Primer2 and Primer3. Primer Design: Design Primer 1 and Primer 2 to amplify Gene1. Settings that I use for VectorNTI or Primer3 are: o 40 < Tm < 65 o 35 < %GC < 60 o 18 < Length < 25 Design Primer 3 and Primer 4 to amplify the tag. If Primer 3 will be the fusion primer, design it to have 25 – 30 bp of homology to the sense strand of the 3’ end of Gene1 + 20 – 25 bp of the sense strand of the 5’ end of the tag. Check your primers well. Primer 3 and Primer 2 should have some reverse complementation, but should not be identical. If you are going to directly transform your fusion product into yeast, design Primer 5 & Primer 6 to have 30 bp of homology to the region of recombination. Osborne, E. 06/2008 Protocol Step 1: Amplify the two separate fragments Set up two parallel PCR reactions to amplify Gene1 and the Tag. Use the Expand Long Template PCR protocol as follows as an example of each reaction: Reagent 1x H2O 40.622 µl 10 x Expand Buffer + 17.5mM MgCl 5 µl 10 mM dNTP's 1 µl 100 μM Primer1 (or 3) 1 µl 100 μM Primer2 (or 4) 1 µl (200 ng/µl) gDNA or (50 ng/µl) Plasmid DNA 1 µl 5 U/μl Expand LT TAQ 0.378 µl 50 Run the PCR for 12 cycles on KO-TAG (below). This is typically not enough to see a band on a gel. However, it is enough to start some products which will be the templates in the Step 2 PCR reaction. Between the two PCR reactions, try to avoid going over 30 cycles total or mutations will start to become a problem. 1. 2. 3. 4. 5. 6. 7. 8. 94C 2:00 min 94C 0:30 min 50C 0:30 min 68C 3:00 min*** Goto Step2 11x 68C 7:00 min 4C for ever END *** This time changes as your expected product length changes. Elongation time = 1min/1kb fragment + 1 min Clean up the fragments. Use a Qiagen PCR Purification Kit or a Qiagen Nucleotide Removal Kit (if your fragments are <200 bp) to clean up the products. Elute in 50 µl EB or H2O. Step2: Fuse the two fragments Using the two products from Step1 as the templates for a single PCR reaction, fuse the two products into one. This reaction will either use Primer 1 and Primer 4 if the products are to be cloned or Primer 5 and Primer6 for direct transformation. Reagent H2O 10 x Expand Buffer + 17.5mM MgCl 10 mM dNTP's 100 μM Primer1 (or 5) 100 μM Primer4 (or 6) Gene1 fragment from Step1 1x 34.622 µl 5 µl 1 µl 1 µl 1 µl 4 µl Osborne, E. 06/2008 TAG fragment from Step1 5 U/μl Expand LT TAQ 4 µl 0.378 µl 50 Run the PCR for 18 cycles on KO-TAG (see above). If the final fragment is to be used in a TOPO TA cloning reaction, add 1µl of AmpliTAQ GOLD TAQ Polymerase and allow the reaction to incubate at 72 C for 10 min. Clean up the fused fragment. Run the entire 50 μl of the fusion reaction on a new 1 % agarose gel + EtBr (made with 1xTAE) in a gel box with fresh 1xTAE. Run the gel to completion (100v, 1hr). Cut the bands and extract the DNA with a Qiagen Gel Extraction Kit. Elute in 50 µl EB or H2O. Future Directions To clone fragments into a vector, use 2 µl of cleaned fusion product in an Invitrogen TOPO TA Cloning Reaction. Tranform 2 µl of the cloning reaction into 50 µl TOP10 e.coli cells. To use fragments for direct transformation into yeast, use all 50 µl of cleaned fusion product in the Manitoba Best Yeast Transformation Protocol. Sequence your results to check for errors. Troubleshooting No band? o o o The most common problem is that there is not adequate starting template in Step1. Try to increase the amount of starting template. If your fusion fragment is small, try to extend the number of cycles beyond the 30cycles recommended, but be aware that mutations will increase as you do so. If your fragments are large and/or stubborn, run the PCR reactions in Step1 for 20 – 30 cycles and clone those products into a TOPO TA vector. Sequence the vectors to find error-free constructs that can be used as starting templates for Step1 amplification, this time with only 12 cycles. This works well especially for amplifying large genes out of gDNA. Too many bands? o Try a higher annealing temp (higher than 50C) in both PCR reactions. Lots of mutations? o Minimize PCR cycles. Only one of the fragments shows up at the end? o This is usually an issue when Step1 amplifies a disproportionate number of one of the fragments compared to the other. Try performing Step1 for 20 – 30 cycles and then running the products on a gel. Chances are, one product is amplifying like crazy, while the other product is not well-represented. Change the number of PCR cycles for each fragment in Step1 so that the two fragments are about equal in the Fusion PCR reaction. Osborne, E. 06/2008 Osborne, E. 06/2008