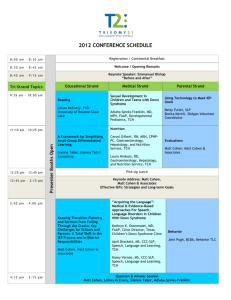
Molecular Diversity New Insights in the Activation of Human
advertisement

Molecular Diversity New Insights in the Activation of Human Cholesterol Esterase to Design Potent Anti-cholesterol Drugs Shalini John, Sundarapandian Thangapandian, Prettina Lazar, Minky Son, Chanin Park, and Keun Woo Lee* Division of Applied Life Science (BK21 Program), Systems and Synthetic Agrobiotech Center (SSAC), Plant Molecular Biology and Biotechnology Research Center (PMBBRC), Research Institute of Natural Science (RINS), Gyeongsang National University (GNU), 501 Jinju-daero, Gazwa-dong, Jinju 660-701, Republic of Korea. *Corresponding author. Tel. +82-55-772-1360; Fax. +82-55-772-1359. E-mail address: kwlee@gnu.ac.kr Supporting Information Text S1 1. Comparison of activation efficiency of different bile salts 1.1. Stability analysis In general the RMSD of the three systems were well converged throughout the simulation. The average backbone RMSD values of hCEase_TCH, hCEase_CHA and hCEase_GCH systems are 0.20, 0.24 and 0.19 nm respectively (Fig. S2A). The potential energy graph has shown that the energy difference between the systems is very less and all the four systems have maintained a stable average value between -12,202 to -12,187 kJ/mol. The Rg was compared to measure the compactness of the structures of the proteins. The Rg value of hCEase_TCH system was maintained at an average value of 2.29 nm. The hCEase_CHA and 1 Molecular Diversity hCEase_GCH systems have shown slightly decreased average values of 2.27 nm and 2.24 nm, respectively. 1.2. Fluctuation analysis The Cα RMSF was measured for the three systems to monitor the fluctuation of each residue in presence of different bile salts. From the RMSF graph it was observed that the areas belonging to the secondary structure elements are more stable. The region in the graph with highest fluctuation belongs to the loops and coils between helices and sheets (Fig. S2B). The fluctuation was high in the loop regions of hCEase_TCH system when compared to the hCEase_CHA that has shown more fluctuation than hCEase_GCH system. The catalytic triad and oxyanion hole residues of hCEase_TCH system was very well stabilized than hCEase_GCH but hCEase_CHA system has shown more fluctuation in this region (Fig. S2C). The RMSF analysis revealed that TCH fluctuated more in the loop regions but stabilized well in the catalytic site region. The hCEase_GCH had considerable stability but the fluctuation of hCEase_CHA was very high in the catalytic site region. 1.3. Structural changes and binding mode analysis The movement of 120-loop, in hCEase_TCH, was in the open conformation than hCEase_CHA, towards the closed conformation, whereas hCEase_GCH was near to the open conformation. It has clearly been observed that the big steroid moiety of TCH was placed in front of the 120-loop and the side chain was nuzzled in the gap between 120- and 450-loop (Fig. 2A). But in case of CHA, due to lack of long side chain the interaction was lost thereby the big steroid moiety cannot move the 120-loop enough to get the open conformation (Fig. 2C). In case of hCEase_GCH, though GCH has lengthy amide group as side chain, due to the lack of SO3- group the strong interaction required to place the big steroid moiety exactly 2 Molecular Diversity opposite to the 120-loop was missing (Fig. 2E). Thereby the conformation of GCH was not in an appropriate position. Hence the active site of hCEase_GCH was largely disturbed. Likewise the remote TCH binds well in the cleft region compared to CHA and GCH as their binding orientations are not favorable and the binding site was largely disturbed. The analyses based on structural changes, H-bond interactions and binding modes revealed that TCH binds the active site in a favorable manner and brought essential conformational changes comparing to binding of CHA and GCH. 1.4. Distance analysis The distance between two key residues, one from the catalytic triad, His435, and another from oxyanion hole, Gly107, was measured throughout the simulation. From the distance graph (Fig. S4) it was observed that hCEase_TCH has maintained an average of 0.527 nm whereas hCEase_CHA and hCEase_GCH has shown decreased and increased average values of 0.487 nm and 0.650 nm, respectively. 1.5. Interaction energy calculations The energy of interaction between individual residues and ligands were calculated for the three different enzyme-ligand complexes to get the comprehensive view of effects of the important residues on the binding affinity of the three different systems. The interaction energy between the residues and the ligand was calculated using the Calculate Interaction Energy protocol available in DS with the default parameters. The important amino acids showing interactions with proximal TCH are Asn117, Asn118, Phe119, Asn121, Asn122, Tyr123, Leu124, Tyr125, Gln440, Lys445, Ala448, Thr449, Pro450, Thr451, Gly452, and Tyr453. The 3α-hydroxyl group of proximal TCH has shown an H-bond interaction with Ile124 and the 7α-hydroxyl group with Thr449. This result was similar to the important 3 Molecular Diversity interaction observed for bCEase. The important amino acids showing interaction with remote TCH molecule are Leu224, Thr352, Thr523, Ile353, Ile399, Trp522, Phe351, Thr354, Tyr526, and Pro396. The contribution of electrostatic and van der Waals interactions to the nonbonded interaction energies of different bile salts TCH, CHA, and GCH that forming direct interactions with active site residues were calculated and summarized (Table S1 and S2). From the result it was observed that the electrostatic energy of the residues Asn117, Phe119, Asn121, Leu124, Lys445, Thr449, Pro450, and Thr451 have major contribution in the ligand binding. The TCH has more satisfactory interaction energies with these important residues comparing to GCH and CHA. Interestingly many of these residues have shown strong Hbond interaction with TCH than the other two ligands. This outcome enhances the importance of 120-loop because the major bonding and non-bonding interacting residues are from the 120-loop. Though CHA and GCH have shown favorable interaction energies their contribution to the electrostatic interaction energy was less than TCH. This favorable interaction energy may be directly related to higher binding affinity of TCH towards hCEase. The van der Waals interactions were the major weakening interactions in terms of CHA molecules that showed a high positive value of 201.868 kcal mol-1 towards Leu124. This is the main reason for its instability and the disturbed mode of binding at the active site. Though the vdw interactions of TCH and GCH have destabilizing effect it was compensated by the more favorable electrostatic contributions. In case of remote bile salt, Ser225, Thr523, Ile399, Trp522, Phe351, Thr354, Tyr526, Pro396, Thr352, and Ile353 residues have shown substantial contributions to the electrostatic interaction energy. The TCH has shown good electrostatic energy when compared to CHA and GCH molecules. The vdw interaction has shown destabilizing effect in CHA but not in TCH and GCH binding because of the beneficial electrostatic interaction. Among all ligands the energy contribution of CHA is less due to the Ile399 residue with 1244.36 kcal mol-1 of vdw interaction. The difference in their 4 Molecular Diversity interaction energy was well explained by the sum of the interaction energies of all residues for TCH, CHA and GCH, -60.067, 257.625 and 3.195 in proximal and -16.123, 1185.293 and 72.822 in remote, respectively. This provides evidence that TCH has strong interaction energy when compare to CHA and GCH. Text S2 2. Importance of both the bile salt binding sites (proximal & remote) 2.1. Overall structural stability In this part, analysis of non-ligated, double and single ligated systems were considered to observe the importance of the presence of both the bile salts and how these bile salts are involved in the activation of the hCEase. The backbone RMSD of these systems was analyzed to scrutinize the behaviors of the fluctuation of each atom with respect to their apoform (hCEase_Apo). The average RMSD values of each system, hCEase_Apo, hCEase_TCH, hCEase_Prx, and hCEase_Rmt, were 0.250, 0.203, 0.204, and 0.205 nm, respectively (Fig. S5A). The apoform of the protein has shown high deviation with an average value of 0.250 nm when compared to the complexed systems. The RMSD results indicated that the ligated systems were more stable than the apoform during the 5 ns MD simulations. The structure of hCEase is mainly comprised of α/β secondary structures which are connected by flexible loops. The flexibility of these loops is directly connected with the activation of the enzyme. The Cα RMSF values of four systems were calculated and analyzed. The overall RMSF distribution was similar for all the four systems. The variable loop residues have shown high flexibility over the residues forming secondary structures (Fig. S5B). Interestingly, the double ligated system, hCEase_TCH, was observed with the largest fluctuations than the other two systems. The binding of proximal TCH is responsible for the 5 Molecular Diversity fluctuation of the residues in the large α/β domain whereas the binding of remote TCH is responsible for the fluctuation of the residues in the small helical domain. Hence the required flexibility of residues of the surface loops is more in double ligated system. The RMSF of catalytic residues was calculated, though hCEase_TCH has shown more flexibility in its surface loops region the catalytic site residues were well stabilized during the MD simulation. The potential energies of the 4 systems were initially fluctuated and stabilized at the value of -1.2 106 kJ/mol during the simulation time. This result suggested that the ligated systems are energetically more stable like the apoform, which is determined crystallographically. The Rg values were calculated for all the four systems to assure their compactness. The ligated systems have shown better values than the non-ligated system. The average Rg values of hCEase_Apo, hCEase_TCH, hCEase_Prx and hCEase_Rmt systems are 2.291, 2.282, 2.279 and 2.264, respectively. The results of stability analyses revealed that all systems were well stabilized during the simulation time. 2.2. Intermolecular hydrogen bonds Similarly, TCH-rmt ligand has shown more number of H-bonds when compared to Rrmt during the simulation time. The TCH-rmt and Rmt-rmt ligands have shown the average H-bond interactions of 4.4 and 3.5 H-bonds, respectively (Fig. S6A). The binding mode of TCH-rmt (Fig. S6B) was very stable due to the strong H-bonds, maximum of 11, with the important residues. While remote alone is present (Rmt-rmt) the binding mode (Fig. S6C) was not stable due to the less H-bond, maximum of 6, interactions. From this result it is very clear that the H-bond interactions are very stable and strong in the double ligated system. A synergistic effect is observed between proximal and remote TCH in the double ligated system where the remote TCH assists the binding of proximal TCH similarly the proximal TCH assists the binding of remote TCH. Therefore TCH-prx and TCH-rmt ligands have shown 6 Molecular Diversity more stable polar and non-polar interactions with hCEase when they are present together. This signifies the importance of the presence of both the bile salts. The hydrophilic and hydrophobic interactions were analyzed using DS, Ligplot, Molegro Virtual Viewer, and Pose View programs. 2.3. Changes in the other loops Apart from 120-loop there are some other important loops such as 70-loop (residues 65-75), 270-loop (residues 270-285), 420-loop (residues 423-433), and 220-loop (residues 222-229) are present around the active site and these loops are called surface loops. The TCH binding influenced the conformational changes of these loops. Like 120-loop the movements of other surface loops were also measured for hCEase_TCH, hCEase_Prx and hCEase_Rmt with respect to hCEase_Apo. The considerable movement was observed in the 70- and 420-loops of all the systems (Fig. S9). The 270-loop is another important loop present near the small domain, the conformational flexibility of this loop plays a crucial role in the accommodation of different substrates. The hydrophobic pocket formed by the 270-loop and small domain can be able to accommodate the fatty acid part of the substrate. This site was proposed as a possible pathway for the product to exit from the catalytic site 1. In the double ligated systems the 270-loop moved 5.2 Å away from the active site similarly in hCEase_Prx system this loop moved 3.0 Å away from the active site whereas in hCEase_Rmt this loop moved 5.8 Å towards the active site. From the observation, it is clear that in the double ligated system the prominent open conformation and in hCEase_Prx the significant open conformation of 270loop were observed. In hCEase_Rmt the closed conformation of 270-loop was observed which is not suitable for a substrate to bind (Fig. S10). The loop conformational changes clearly explain that if both the TCH (proximal and remote) are present the surface loops have shown favorable conformational flexibility. The appreciable conformational flexibility of the 7 Molecular Diversity loops was observed while prx alone binding at its site but this flexibility was very less in the rmt alone system. This means both the bile salts are mandatory for the flexibility of surface loops but particularly prx involved more. Thereby binding of proximal TCH not only shows great influence on the 120-loop but also on the 270-loop. Hence binding of both the TCH stabilized the protein and adjusted all the surface loops thereby made the active site big enough to accommodate the substrate molecules. 2.4. Changes in the catalytic site The distance analysis was carried out to observe the changes happened at the catalytic site of the protein during MD simulations. The distance between two key residues Gly107 and His435 from the oxyanion hole and the catalytic triad, respectively, was calculated. These residues present in the direction extremely opposite to each other in the catalytic site. Thus the calculation of distance between them was done to ascertain the changes happened due to the presence of TCH. Interestingly, in the double ligated system the distance was less with an average value of 0.527 nm. In case of single ligated systems, hCEase_Prx and hCEase_Rmt, the distance was large with an average value of 0.660 and 0.611 nm respectively similar to the value of hCEase_Apo which has shown an average value of 0.569 nm (Fig. S11). The distance analysis revealed that the required distance between the key residues for an effective hydrolysis process in the catalytic site were maintained well in the double ligated system. 8 Molecular Diversity Text S3 3. Importance of 7α-hydroxyl group in TCH 3.1. Overall system stability The backbone RMSD calculated for both the systems (Fig. S13) were well converged throughout the simulation with an average of 0.203 and 0.227 nm (Fig. S13A). The potential energies and the Rg of the systems were measured and it was observed that both the systems were equilibrated well and stable throughout the simulation. The Cα RMSF revealed that the absence of 7α-OH group greatly influence the flexibility of the protein (Fig. S13B). When 7αOH is present the flexibility of the surface loops was high but in absence of 7α-OH the fluctuation reduced considerably. The distance between the key residues Gly107 and His435 was measured to check the size of the catalytic site (Fig. S14). Interestingly, the increase in the distance was observed from 0.527 to 0.681 nm when 7α-OH is not present thereby leads to the disruption in the catalytic process. The basic analysis results revealed that the flexibility of the surface loops required for the activation of the enzyme was reduced and it also disturbed the catalytic machinery due to the absence of 7α-OH. 3.2. Analysis of intermolecular hydrogen bonds Similarly the 7α-OH of TCH-rmt has formed strong H-bond with Leu224. It also aids TCH to have interaction with Trp522, Lys231, Ile353, Thr354, and Tyr526. In case of TCH_XOHrmt which did not show interaction with Leu224, Lys231, and Trp522 has shown strong interaction with Tyr526, Thr354, and Ile353 (Fig. S16). Throughout the MD simulation, strong and stable H-bonds were maintained by TCH-rmt, maximum of 11 and an average of 4.4, whereas TCH_XOH-rmt has shown weak interactions, maximum of 7 and an average of 3.6. From this H-bond interaction analysis it has become very clear that missing a 7α-OH 9 Molecular Diversity group exerted a huge difference in the protein-ligand binding. This result provided the molecular explanation for the fact that the key interaction of 7α-OH group is very important for the activation of hCEase. The lack of stable H-bond interactions was mainly due to the absence of 7α-OH group. Though the TCH_XOH forms minimum number of H-bonds the necessary interactions required for the activation of enzyme was missing. As a result the active site became collapsed and not suitable for a substrate or inhibitor to bind. 3.3. Interaction energetics The interaction energies of the important residues in the TCH and TCH_XOH system were compared. The results showed the favorable electrostatic interaction energy with the important residues such as Ala117, Asn118, Asn121, Asn122, Tyr123, Leu124, Gln440, Lys445, Thr449, Pro450, Thr451, Gly452, and Tyr453. In hCEase_TCH, the proximal site residues such as Phe119, Asn121, Thr449, Pro450 and Thr451 has shown strong electrostatic interaction of -6.348, -6.916, -4-869, -4.247 and -8.881 kcal mol-1 with the ligand whereas the same residues have shown weak electrostatic interaction 1.597, -1.770, -0.300, -0.521 and 2.054 kcal mol-1 with the TCH_XOH (Table S3). Similarly, favorable interaction was observed for TCH in the remote site (Table S4). The total interaction energy for all residues of TCH and TCH_XOH in proximal and remote sites are -60.067, -53.210, and -16.123, 72.822 kcal mol-1 respectively. 10 Molecular Diversity Table S1 Non-bonded interaction energies in kcal/mol between proximal bile salts and protein of hCEase_TCH, hCEase_CHA, hCEase_GCH systems Residue TCH CHA GCH Coulomb Vdw Coulomb Vdw Coulomb Vdw Ala117 3.406 -2.312 3.693 3.788 -3.346 -0.118 Asn118 -1.134 -1.599 -1.249 -1.361 -0.680 -0.168 Phe119 -6.348 -2.921 -1.408 32.862 -2.076 -4.131 Leu120 -2.125 -0.105 -1.965 -0.275 1.644 43.199 Asn121 -6.916 -1.337 -3.779 -0.152 -10.282 -0.577 Asn122 0.162 -7.588 1.566 -2.894 -1.218 -1.224 Tyr123 3.404 -3.719 -2.589 -0.700 1.993 -0.258 Leu124 -0.893 1.273 -0.827 201.868 0.510 -1.224 Tyr125 2.411 -0.240 -0.730 -0.065 -0.018 -0.091 Gln440 0.141 -0.305 0.158 -2.094 4.921 -1.526 Lys445 11.877 -1.951 -0.851 -1.773 -10.749 -2.565 Ala448 2.469 -0.371 0.076 -0.179 -6.051 -0.256 Thr449 -4.869 -5.387 2.647 41.523 -1.464 -3.932 11 Molecular Diversity Pro450 -4.247 -0.562 -1.195 -0.207 -2.422 -1.209 Thr451 -8.881 9.597 -1.794 -2.944 -6.618 -5.803 Gly452 1.309 -0.475 -0.092 1.027 -2.803 -0.365 Tyr453 0.056 -2.639 -4.110 -4.638 1.933 -0.786 12 Molecular Diversity Table S2 Non-bonded interaction energies in kcal/mol between remote bile salts and protein of hCEase_TCH, hCEase_CHA, hCEase_GCH systems Residue TCH CHA GCH Coulomb Vdw Coulomb Vdw Coulomb Vdw Leu224 3.392 -2.523 -0.626 -2.055 -3.040 -2.708 Ser225 4.507 -0.328 2.666 -0.736 -5.624 -0.465 Phe351 -2.906 -2.131 -0.208 -3.333 2.923 -2.265 Thr352 -1.813 -1.417 2.965 -1.116 1.175 -1.774 Ile353 -2.015 -3.010 1.794 -2.151 -1.796 -1.236 Thr354 -3.070 16.543 -9.399 5.189 -2.886 -0.497 Pro396 -0.073 -2.452 1.545 -3.133 -3.118 -1.932 Ile399 -0.569 -2.414 1.282 1244.360 2.028 -4.125 Trp522 -0.710 -2.691 0.955 -4.742 1.054 -7.408 Thr523 -4.406 1.289 1.658 -0.776 10.953 -2.423 Tyr526 -6.170 5.508 -4.079 -3.641 -0.480 -5.091 13 Molecular Diversity Table S3 Non-bonded Interaction Energy values between proximal hCEase_XOH and hCEase_TCH Residue TCH TCH-XOH Coulomb Vdw Coulomb Vdw Ala117 3.406 -2.312 -3.068 -0.466 Asn118 -1.134 -1.599 -1.729 -2.258 Phe119 -6.348 -2.921 1.597 14.147 Asn121 -6.916 -1.337 -1.770 -0.190 Leu120 -2.125 -0.105 -1.144 -0.315 Asn122 0.162 -7.588 -7.658 -3.786 Tyr123 3.404 -3.719 -1.990 -0.673 Leu124 -0.893 1.273 -5.815 -1.511 Tyr125 2.411 -0.240 4.304 -0.309 Gln440 0.141 -0.305 -0.664 0.221 Lys445 11.877 -1.951 -7.749 -1.584 Ala448 2.469 -0.371 1.596 -0.077 Thr449 -4.869 -5.387 -0.300 -2.474 14 Molecular Diversity Pro450 -4.247 -0.562 -0.521 -0.187 Thr451 -8.881 9.597 -2.054 7.242 Gly452 1.309 -0.475 -0.534 -1.286 Tyr453 0.056 -2.639 -0.198 -2.296 15 Molecular Diversity Table S4 Non-bonded interaction energy values between remote hCEase_XOH and hCEase_TCH Residue TCH TCH-XOH Coulomb Vdw Coulomb Vdw Leu224 3.392 -2.523 -6.170 10.589 Ser225 4.507 -0.328 6.549 -0.298 Phe351 -2.906 -2.131 1.262 6.127 Thr352 -1.813 -1.417 -1.824 -1.644 Ile353 -2.015 -3.010 -4.437 -2.969 Thr354 -3.070 16.543 -6.016 2.494 Pro396 -0.073 -2.452 -0.177 -2.216 Ile399 -0.569 -2.414 1.946 122.338 Trp522 -0.710 -2.691 2.749 -4.284 Thr523 -4.406 1.289 0.508 -1.753 Tyr526 -6.170 5.508 -6.089 -2.950 16 Molecular Diversity Fig. S1 Non-ligated and different ligated systems used in MD simulations. The bile salt binding sites are shown in black circles 17 Molecular Diversity Fig. S2 Basic analysis plots. A) The backbone RMSD graph was plotted to check the overall stability of hCEase_TCH, hCEase_CHA and hCEase_GCH systems. The Cα-RMSF graph for B) all protein residues C) catalytic site residues. The more fluctuating and catalytic residues are highlighted in the RMSF plot 18 Molecular Diversity Fig. S3 Structural changes in the surface loop regions. The conformational changes of surface loops were observed and compared between hCEase_TCH and (A) hCEase_CHA (B) and hCEase_GCH systems. The distance between the loops were measured in Armstrong and represented in black dotted lines. The thick arrows pointing the distance of the two major 120 and 270 loops 19 Molecular Diversity Fig. S4 Distances between the catalytic residues during MD simulation. The distances between two important catalytic site residues, Gly107 and His435, located at the active site were measured in hCEase_TCH, hCEase_CHA, and hCEase_GCH systems throughout the MD simulations 20 Molecular Diversity Fig. S5 Stability analyses of four systems. The A) backbone RMSD and B) Cα-RMSF plot of hCEase_Apo, hCEase_TCH, hCEase_Prx and hCEase_Rmt systems 21 Molecular Diversity Fig. S6 H-bond interactions and binding modes of TCH at the remote site. (A) The number of H-bond interactions between protein and remote ligands of hCEase_TCH and hCEase_Rmt systems throughout the MD simulations was measured. B) and C) The binding modes of TCH at the active site of TCH-rmt and Rmt-rmt systems respectively. The key interacting residues were shown in white and green color sticks whereas the ligands were shown in deep teal color sticks. The H-bond interactions are shown in black dashed lines 22 Molecular Diversity Fig. S7 Structure overlay of hCEase and bCEase. Comparison of MD simulated hCEase (gray color) with the crystal structure of bCEase (green color). The TCH was shown in stick form and the surface loops around the active site are labeled 23 Molecular Diversity Fig. S8 Comparison of 120-loop movement. Distances between the residue Leu120 present in the 120-loop of hCEase_Apo and hCEase_TCH, hCEase_Prx, hCEase_Rmt, respectively, was measured and provided in Armstrong scale after 5 ns MD simulations. The distance was shown in black dashed lines Fig. S9 Conformational changes of surface loops. The conformational changes of surface loops were observed between hCEase_Apo and A) hCEase_TCH, B) hCEase_Prx, and (C) hCEase_Rmt systems. The distance were measured and represented in black dashed lines 24 Molecular Diversity Fig. S10 Conformational changes of 270-loop. Conformational changes of 270-loop were observed between A) hCEase_TCH and hCEase_Prx and B) hCEase_TCH and hCEase_Rmt systems. The important residue is shown in stick form Fig. S11 Distances between the key residues in the catalytic site. The distances between two important catalytic site residues, Gly107 and His435, located at the active site were measured in hCEase_Apo, hCEase_TCH, hCEase_Prx, and hCEase_Rmt systems throughout the MD simulations 25 Molecular Diversity Fig. S12 The 2D structures of TCH and TCH_XOH. The position of 7α-OH group is highlighted in green color for TCH and red color for TCH_XOH 26 Molecular Diversity Fig. S13 Stability analyses plot. The A) RMSD and B) RMSF analyses plots for, hCEase_TCH and hCEase_XOH systems, respectively. The most fluctuating residues were labeled in the RMSF plot 27 Molecular Diversity Fig. S14 Distances between Gly107 and His435 plot. The distances between two important catalytic site residues, Gly107 and His435, at the active site of hCEase_TCH (grey) and hCEase_XOH (pink) were measured throughout the MD simulations. The average distances for both the systems are graph. 28 given in the Molecular Diversity Fig. S15 H-bond interaction analysis. The H-bond interactions between protein and ligand in the A) proximal and B) remote sites were measured during 5 ns MD simulation 29 Molecular Diversity Fig. S16 Binding modes of TCH and TCH_XOH at the remote site. The binding modes and H-bond interactions between protein and ligand A) TCH and B) TCH_XOH are shown in deep teal color and lime color stick forms. The missing 7α-OH group region is highlighted in red circles. The H-bond interactions are shown in black dashed lines 30 Molecular Diversity Fig. S17 Conformational changes of the surface loops. Comparison of conformational changes of the surface loops between hCEase_TCH and hCEase_XOH systems 31