Working Experience
advertisement

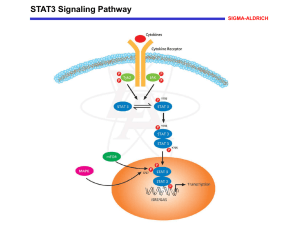
Curriculum Vitae Jianping Lin Address 1001 Arcadia Ave. #2 Arcadia, CA 91007 Email: jianpingl@gmail.com Phone: 626-241-6163 Education 2000.8 – 2005.8 Ph.D. Advisor: Duke University, Durham, NC in Theoretical Biophysics and Biochemistry Prof. David N. Beratan 1997.8– 2000.7 M.S. Advisor: Beijing University, Beijing, China in Polymer Chemistry Prof. Gaoyuan Wei 1992.8 – 1997.7 B.S. Beijing University, Beijing, China in Chemistry Advisor: Prof. Gaoyuan Wei Working Experience 2006.3Research Fellow City of Hope National medical Center 2005.9-2006.2 Research associate Duke University 2001.9-2005.8 Research Assistant Duke University 2001.5-2001.8 Research Assistant University of Pittsburgh 2000.8-2001.4 Teaching Assistant University of Pittsburgh 1997.8-2000.7 Research Assistant Beijing University 1992.8-1997.7 Undergraduate Research Beijing University Techniques Molecular Dynamics, essential dynamics Brownian Dynamics, Monte Carlo Gaussian Network Model Virtual Ligand Screening and post analysis Software Skills Gaussian 98 UHBD, Sybyl NAMD, VMD Maestro 8 (including GLIDE, PRIME, INDUCE-FIT DOCK) Workshop Attended • Summer school on theoretical and computational biophysics: Computational approaches for simulation of biological systems (June 2-13, 2003) Organizer: Professor Klaus Schulten, Beckman Institute, University of Illinois • Research workshop and summer school on theory and computation in molecular biological physics (August 9-20, 2004) Organizer: Professor Charles L Brooks III, Department of Molecular Biology, the Scripps Research Institute. Curriculum Vitae Research Experiences Molecular dynamics simulations of insertion of chemically modified DNA nanostructures into water-chloroform interface DNA based 2D and 3D arrays have been used as templates for synthesis of functional polymers and proteins. Hydrophobic or amphiphilic DNA arrays would be useful for the synthesis of hydrophobic molecules. The objective of this study is to design modified amphiphilic double crossover DX-DNA molecule that would insert into waterchloroform interface thus showing amphiphilic character. Since experiments for such design are tedious, we have used molecular dynamics simulations to identify and optimize the functional groups to modify the DNA backbone, that would enable insertion into the water-chloroform interface, prior to synthesis. By methylating the phosphates of the backbone, to make phosphonates, combined with placing a benzyl group at the 2’ position of the deoxyribose rings in the backbone, we observed that the simple B-DNA structure was able to insert into the water-chloroform interface. We find that the transfer free energy of the methylated benzylated DNA is better than either just methylated or benzylated DNA. The driving force for this insertion comes from entropic contribution to the free energy and the favorable van der Waals interaction of the chloroform molecules with the methyl and benzyl groups of the DNA. (Accepted by Biophysical Journal, 2008). Molecular dynamics simulations of the conformational changes in Signal Transducers and Activators of Transcription, Stat1 and Stat3 All Signal Transducers and Activators of Transcription (STAT) factors are a family of cytoplasmic transcription factors that mediate the signal response to cytokines, growth factors, and hormonal factors. The activation of Stat3, a member of the STAT family, has been found to be elevated in a large number of diverse human cancers. It is pertinent to understand the dynamics of the dimer interface to enable Stat3 dimer inhibitor design. To this end, we performed 50 ns MD simulations for Stat3 dimer, and its closely related member of the family, Stat1 dimer in explicit water. We observed a large scale domain motion in Stat3 dimer while the structure of the monomer remains intact. The driving force for this conformational change is enhanced binding of the Stat3 dimer to the DNA, thereby promoting transcription. Our model shows that the carboxy terminus of one monomer wraps around the core of the SH2 domain of the other monomer, and this region that makes up the dimer interface remains intact during the dynamics. Water diffuses into a cavity under this interface thus expanding a pre-existing cavity that gets gated and closed by the loops in the SH2 domain. This cavity serves as a potential binding pocket for inhibitor design. (Submitted to J. Am. Chem. Soc. May, 2008) Design of inhibitors for signal transduction and activator of transcription (STAT3) by targeting new binding sites We first performed 2.6 ns molecular dynamics on Stat3 crystal structure. For the equilibrated structure, we performed virtual ligand screen for the binding site right under Jianping Lin 2 Curriculum Vitae the crossover (This is the first research to target this site) between two c-terminal of STAT3 dimer from Sequeonom database. We pick the top10% ligands from GLIDE score. Using our own filter strategy, we narrow down the number of ligands to 437. Among the 437 ligands, our collaborators pick 52 ligands to test in the inhibition of DNA binding for STAT3 and found one compound can inhibit the binding at IC50 <= 30 μM in EMSA (electrophoretic mobility shift assay). And it is proven to have specificity for STAT3 instead of Stat1 and no toxicity to normal cell. This compound could be anticancer drug with specificity by targeting STAT protein. The results will be included into a manuscript in preparation (to be submitted to Cell) and a patent to be applied. The solvent effect on inter-protein electron transfer We explore the distance dependence of electronic coupling between redox cofactors, mediated in part by water, as two redox proteins approach. Rather than describe the ET rate decay with a single exponential parameter, we employ explicit electronic structure calculations and find three distinct tunneling mediation regimes: a conventional protein-mediated regime near protein-protein contact, a “structured water” regime with soft distance dependence for small protein-protein gaps, and a bulk water regime with a rapidly decaying coupling at larger distance. We use this analysis to predict the rate for cytochrome b5 electron self-exchange. These results are published in Science. Simulation of electron transfer between cytochrome c2 and the bacterial photosynthetic reaction center In bacterial photosynthesis, electron transfer from a cytochrome to the special pair of the reaction center completes the cycle of converting light into chemical energy. We used Brownian dynamics to compute the second-order electron transfer rate, between cytochrome c2 and bacterial photosynthetic reaction center. The results compare well with the experimental kinetic results. We also predict that the second-order electron transfer rate decreases with increasing ionic strength, a characteristic of electrostatically controlled docking. We predict that double mutations of the system can switch the mechanism between activation & diffusion control. These results are published in J. Phys. Chem. B. Simulation of tunneling through flexible molecules in scanning tunneling microscopy Since electron-tunneling interactions are exponentially sensitive to geometry changes, thermal fluctuations are expected to have a large influence on roomtemperature tunneling currents and scanning tunneling microscopy (STM) images. Our analysis of STM currents follows the approach of Marcus and co-workers. We use this strategy to explore current variation among specific molecular conformations from which we compute the conformationally averaged currents for several fixed tip-to-substrate distances. We simulate the process of pulling a octanedithiol molecule from a “random coil” to a fully extended form. We find that the tunneling current is expected to have a strong conformational dependence. The predicted tunneling currents in the saturated structures decrease approximately exponentially with distance as the structure are extended. This observation is consistent with a rule that “strong conformers win”. The results are published in J. Phys. Chem. A. The cooperative motion of ATPase Jianping Lin 3 Curriculum Vitae Adenosine triphosphate (ATP) chemical-bond energy drives large-scale conformational changes in proteins. H+-ATPase pumps protons in the plasma membrane to maintain the intracellular pH and membrane potential. To understand the molecular mechanism of proton pumping, we are using a coarse-grained strategy (normal mode analysis with a Gaussian network model) and essential dynamics to analyze the cooperative motion of the ATPase. We use molecular dynamics and Brownian dynamics methods to explore H+ transport through the ion channel. Publications Jianping Lin; Nadrian C. Seeman; Nagarajan Vaidehi “Molecular dynamics simulations of insertion of chemically modified DNA nanostructures into waterchloroform interface” Accepted by Biophysical Journal in April, 2008. James C. Sung; Jianping Lin Diamond Nanotechnology: Synthesis and applications Pan Stanford Publishing, in press. Jianping Lin; Nagarajan Vaidehi “Molecular dynamics simulations of the conformational changes in Signal Transducers and Activators of Transcription, Stat1 and Stat3” Submitted to J. Am. Chem. Soc. In May, 2008. Jianping Lin; Ralf Buettner; Richard Jove; Nagarajan; et al. “A novel inhibitor for transduction and activator of transcription (STAT3) by targeting new binding site“ In preparation ( to be submitted to Cell, 2008). Jianping Lin; Ilya A. Balabin; David N. Beratan “The Nature of Aqueous Tunneling Pathways between Electron-transfer Proteins” Science 2005, 310,1311-1313. related news: “Water can ease electron transfer between proteins” Chemical & Engineering News November 28, 2005. Page 11. Jianping Lin; David N. Beratan “Simulation of Electron transfer between Cytochrome c2 and the Bacterial Photosynthetic Reaction Center: Brownian Dynamics Analysis of the Native Proteins and Double Mutants.” J. Phys. Chem. B 2005, 109, 7529-7534. Jianping Lin; David N. Beratan "Tunneling while Pulling: The Dependence of Tunneling Current on End-to-End Distance in a Flexible Molecule." J. Phys. Chem. A 2004, 108, 5655-5661. Spiros S. Skourtis; Jianping Lin; David N. Beratan “The Effects of Bridge Motion on Electron Transfer Reactions Mediated by Tunneling” Chapter 18 in Modern Methods for Theoretical Physical Chemistry of Biopolymers. Edited By E. B. Starikov; S. Tanaka; J. P. Lewis, Elsevier Press, June 2006. Awards Outstanding Student Award of Beijing University (1995-1996) Outstanding Graduate Award of Beijing University (1998-1999) Presentation • Jianping Lin; Iyla A. Balabin; David N. Beratan “Interprotein Electron Transfer through Aqueous Pathways” APS meeting in Los Angeles, March, 2005. Jianping Lin 4