here
advertisement

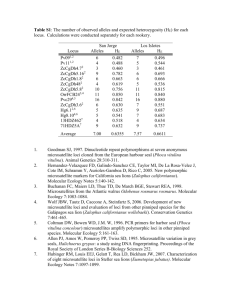
Supplemental Information Appendix 1. Isolation and characterization of the 14 Sitobion miscanthi microsatellite loci Sitobion miscanthi microsatellites were cloned by ACCW and PS at Macquarie University in 1996. DNA was extracted and pooled from multiple parthenogenetic females of S. miscanthi (Clone Sm80, chromosomal race 2n = 18, microsatellite genotype Sm18.0, see Sunnucks et al. 1996) by a salting-out protocol (Sunnucks & Hales 1996). Digestion of DNA for cloning was performed in One-phor-all buffer (Pharmacia), with 4 mM spermidine trichloride using AluI, RsaI and HaeIII (selected because they are four-base cutters that produce blunt ends). Microsatellites were cloned and screened using commercial copolymers, poly(AC).poly(TG) and poly(TC).poly(AG) (Pharmacia) largely following Taylor et al. (1994), except that ligated DNA was transformed into NM522 electrocompetent cells (Sambrook et al. 1989 and Bio-Rad Gene Pulser protocol). In addition, ligations were found to be much more efficient if incubated for 48 hours. All sequencing was performed manually using a Sequenase Version 2.0 kit (United States Biochemical). Primers were designed from the flanking sequences of microsatellite-containing clones using PRIMER0.5 (Lincoln et al. 1991). Approximately 20,000 recombinant S. miscanthi colonies were screened. Sequences were obtained from 25 microsatellite-containing plasmids. Primers were designed from the 17 suitable sequences. Three primer pairs were abandoned because they amplified poorly or not at all. Ten of the 17 loci contained pure CA repeats ranging in length from 11 to 30 repeat units. Three loci contained GA repeats: S23 and one of the abandoned loci had pure repeats of 14 and 25 units respectively, and another abandoned locus had an impure GA repeat. Although not probed for, one impure trinucleotide repeat (S25) and two TA dinucleotide repeats (S35 and S45) were also isolated. Two loci contained compound repeats (S17b, S43ii) and S12 contained an impure (CA)8 (Table 1). Of the 14 useable loci reported, four were identified as X-linked (S10, S17b, S43ii and S49) five as autosomal (S16b, S19, S23, S24 and S30) and the remaining five loci of undetermined location (S9, S12, S25, S35 and S45) (Table 1). Excluding the two loci that appeared to be length-invariant (S12 and S35), the number of alleles per locus ranged from two to 15 over the 11 - 13 genotypes representative of Australian and New Zealand S. miscanthi and S. near fragariae (Table 1). The mean number of alleles per locus was 7. Heterozygosity at these loci ranged from 0.09 to 1.00 (Table 1). Microsatellite PCRs were carried out as described in Sunnucks et al. (1996). PCR cycling conditions (‘touchdown’ programs) were as follows: initial denaturation 94 C 2 mins, followed by one cycle of v C 30 sec, 72 C 45 sec, 94 C 15 sec; one cycle of w C 30 sec, 72 C 45 sec, 94 C 15 sec; one cycle of x C 30 sec, 72 C 45 sec, 94 C 15 sec; one cycle of y C 30 sec, 72 C 45 sec, 94 C 15 sec; 30 cycles of z C 30 sec, 72 C 45 sec, 94 C 15 sec; final extension 72 C for 2 mins. For PCR program PMS1: v = 62, w = 61, x = 59, y = 57 and z = 55; PMS2: v = 55, w = 53, x = 51, y = 49 and z = 47. For PCR program AMS1 the first four extension cycles (temps v-y) were each cycled twice through and the z temperature cycle repeated 28 times. For AMS1: v = 65, w = 64, x = 63, y = 61 and z = 60. Reference DNA can be obtained from the authors where confirmation of allelic length identity is necessary. Isolation and characterization of Sitobion avenae microsatellite loci Microsatellites were cloned by JCS and AE at INRA Jouy-en-Josas in 1996 from a single clone of Sitobion avenae. Microsatellite isolation followed the procedure of Estoup et al. (1998). Briefly, DNA was extracted from 100 aphids using the saltingout method (Sunnucks & Hales 1996) and a partial library was constructed after complete digestion of DNA by Sau3AI. Fragments of 500-1000 base pairs were ligated into pUC18 (Pharmacia Biotech) and transformed using XL1-blue competent cells (Stratagene). A set of digoxigenine-labeled probes [(TG)10, (CAC)5CA, (TGTA)6TG] was used to screen the size-selected library. Approximately 12,000 recombinant S. avenae colonies were screened. Sequences were obtained from 20 positive clones. Primers were designed from the five suitable sequences using PRIMER3 (Rozen & Skaletsky 2000). One locus was abandoned because of amplification failure even after redesigning primers. Only one of the four loci (S5.L) contained a pure CA repeat. Locus S4.Σ, for which primer sequences have already been published (Simon et al. 1999), contained an interrupted AC repeat 50 nucleotides long. The remaining two loci contained compound repeats (Table 1). The number of alleles per microsatellite locus ranged from four to six across populations of S. avenae in Western France (Table 1). The mean number of alleles per locus was 4.8. Observed heterozygosity at these loci ranged from 0.51 to 0.80 (Table 1). Microsatellite loci were amplified in a final volume of 15 l using 3 pmol of each primer, 50 M of each dNTP, 1.5 l of 10X Mg++ free reaction buffer (500 mM KCl, 100 mM Tris-HCl pH 9.0 and 1.0% Triton X-100), 2 mM of MgCl2, 0.5 U of Taq DNA polymerase (Promega) and 1 l of DNA (5 ng). PCRs were performed on a Hybaid thermocycler with the following program. One denaturation step at 94 °C for 2 min followed by 35 cycles with a denaturation step at 94 °C for 20 sec, 20 sec at a locus-specific annealing temperature (Table 1) and an elongation step at 72 °C for 20 sec; then a final extension at 72 °C for 2 min. Amplification products were resolved on a 6% polyacrylamide urea electrophoresis gel and visualized after silver nitrate staining as described by Budowle et al. (1991). Allele sizes were assigned using the sequence of the pGEM-3Zf(+) vector (Promega). Isolation of Myzus persicae microsatellite loci The four M. persicae microsatellite loci described in Table 1 were cloned by Gavin Malarky and RLB in the Department of Entomology, The Natural History Museum London in 1997. Sloane et al. (2001) who identified the linkage groups of sixteen microsatellite loci in M. persicae, including the four described here, found myz3 and myz25 to be X-linked and myz2 and myz9 to belong to the same autosomal linkage group (i). Amplification of the myz loci follows Sunnucks et al. (1996) and details of PCR cycling conditions for programs PMS1, PMS2 and AMS1 are the same as those described above for the amplification of S. miscanthi microsatellite loci. These loci have been extensively used in previous studies of the genetics and population biology of the M. persicae complex (Hales et al. 2000; Sloane et al. 2001; Hales et al. 2002a, 2002b; Wilson et al. 2002; Guillemaud et al. 2003; Wilson et al. 2003) but to date have only been employed in one cross-species amplification study. Locus myz2 has been demonstrated by CCF and CCR to amplify in two species of the genus Neuquenaphis. In 170 N. staryi individuals they found three alleles between 272 and 276 base pairs in size and in 81 N. edwardsi individuals there were three alleles between 197 and 200 base pairs. It is worth noting that the Sloane et al. (2001) M. persicae loci for which we have investigated cross-species amplification in this paper (Table 2, loci M35, M37, M40, M49, M55, M62, M63, M77, M86 and M107) have been demonstrated to amplify in a range of species in the tribes Aphidini and Macrosiphini and the distantly related subfamily Neuquenaphidinae. This, together with the fact that two of the four myz loci are X-linked (X-linked loci appear to amplify more successfully than autosomal loci in cross-species applications, see discussion at the end of the main paper), suggests that inclusion of these four loci in cross-species amplification screening in non-target species may yield useable loci. Non-target amplification assay Loci were assayed for amplification in non-target species using DNA from one to several individuals per non-target species (depending DNA/tissue availability) under standard target-species PCR conditions and resolved on agarose gel. Each assay included, for each locus, at least one target species positive control and a no DNA negative control. The results of this assay were scored as positive if a PCR product of similar size to the target species was present and the no DNA control was blank, negative if non-target samples were blank, the target species positive control amplified and the no DNA control was blank and equivocal if the locus amplified in non-target species weakly or irregularly across a number of individuals. For fourteen non-target species, we extensively investigated non-target species amplification and polymorphism in a greater number of individuals using high resolution denatured polyacrylamide gel electrophoresis (see Supplementary Information Table 2).