Assembling a genome parts list for the immune system
advertisement

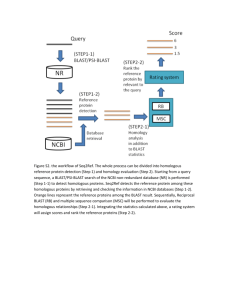
Supplementary Information for Fahrer et al. A genomic view of immunology. Aude M. Fahrer, J. Fernando Bazan1, Peter Papathanasiou, Keats A. Nelms, Christopher C. Goodnow. Medical Genome Centre, John Curtin School of Medical Research, Australian National University, Canberra, Australia, and 1Dept. of Molecular Biology, DNAX Research Institute, CA, USA. Supplementary information. Protein homology searches: Several excellent databases are freely available over the internet to search for protein homologues. These include NCBI [http://www.ncbi.nlm.nih.gov/BLAST/], Interpro [http://www.ebi.ac.uk/interpro/], SMART [http://smart.embl-heidelberg.de/], CLUSTR [http://www.ebi.ac.uk/clustr/] and BIOSPACE [http://biospace.stanford.edu/]. Problems arise, however when trying to interrogate the human genome sequence. Most of the contigs are heavily fragmented; broken into, for example, 15-20 pieces of 1kb, which have been randomly ordered, and separated by runs of “N”s. This leads to problems when trying to predict open reading frames and virtual transcribed sequences (VTS). Most efforts at trying to order the genome, and in predicting VTS are being done by private companies, and are not freely or even publicly available. An exception is the University of California, Santa Cruz effort which is publicly available [http://genome.ucsc.edu/] but can’t be directly queried by homology searches. For this reason, the databases compiled by the Sanger Centre (discussed below), which incorporate novel predicted peptides from the human genome sequence, as well as known proteins turned out to be exceptionally useful. Some examples of different types of homology searches, in increasing order of difficulty, are shown. The first two are accessible to the averagely computer-literate immunologist. The last requires much more specialised bioinformatics. 1. Looking for protein with previously identified motifs. The TNFR family is an extremely important group of molecules which can control either the proliferation or the apoptosis of lymphocytes. Despite having low overall homology (20-25%), the TNFR family is defined by conserved cysteine residues in the extracellular ligand binding domain 1. The family can be divided into two groups based on the presence or absence of a death domain 2. Typing “TNFR” into the Interpro search site [http://www.ebi.ac.uk/interpro/] allows you to retrieve accession IPR001368 for the TNFR/NGFR family cysteine-rich region domain. If this is used to search the Sanger centre protein tables [http://www.sanger.ac.uk/Users/lmc/Ensembl4/collapsed.families/html/index.html], 31 human proteins containing this domain are found. These were compared against the known TNFR family members enumerated in a recent review 2. Multiple nomenclatures for each protein member were quickly unravelled using NCBI’s Online Mendelian Inheritance in Man (OMIM) website [http://www.ncbi.nlm.nih.gov/]. It was found that 21 of the 22 known TNFR family members were represented, the exception being TNFR superfamily member 18. Several members were represented two or three times. Five proteins in the list did not immediately match known TNFR family proteins. One of these was the very recently published TAJ 3. The other four were potentially novel proteins with IGI_ accession numbers. By performing BLAST searches [http://www.ncbi.nlm.nih.gov/blast/blast.cgi?Jform=1] on these four proteins, it was found that two corresponded closely to known TNFR family members OPG and DR5. IGI_M1_ctg13384_53 was similar (but not identical) to OX40, and IGI_M1_ ctg13980_78 was similar, but only over its N-terminal half, to CD30. Thus, over 95% of the known TNFR family members were represented in these tables, and two potentially new and uncharacterised members of the family were rapidly pinpointed. b) If your protein family of interest is not represented by an Interpro domain: The CD80/CD86 molecules (also known as B7.1/B7.2) are members of the immunoglobulin superfamily and are expressed on antigen presenting cells. They share 26% identity and 46% homology at the amino acid level. They both interact with CD28 and CD152 (CTLA-4) molecules on the surface of T cells to transmit either costimulatory or inhibitory signals to the T cell. Recently two new homologs of CD80 and CD86 have been identified 4: ICOSL which binds to the T cell receptor ICOS; and B7H1 which binds to the inhibitory receptor PD-1 5. Crosslinking of ICOS is also costimulatory for T cells, but leads to a different type of T cell response than co-stimulation through CD28. Clearly then, the CD80/CD86 family of proteins is critical for modulating T cell responses. Can we find other members of the family? Since these proteins are not represented in Interpro, we looked for new members using BLAST. Most useful in this case is the position specific iterated PSI-BLAST search 6. This allows searching based on the similarity to several proteins at once, resulting in much greater sensitivity in identifying proteins with weak, but potentially biologically significant, similarities. PSI-BLAST can be accessed through NCBI [http://www.ncbi.nlm.nih.gov/blast/psiblast.cgi], and can be used to search the Genbank databases. Unfortunately, novel peptides predicted from the human genome sequence are not yet available for searching. With somewhat more effort, PSI-BLAST can also be downloaded and used to search other databases. We chose this second option, and used the International Protein Index (version 1) compiled by Ewan Birney at the Sanger Centre [http://www.ensembl.org/IPI/]. We downloaded the expanded peptide database [ftp://ftp.sanger.ac.uk/pub/birney/humanproteome]. By first doing a normal BLAST search against CD80, we found that the first 5 matches (with E values) were: CD80 (e-161); CD86 (5e-12); 2 proteins corresponding to ICOSL (both 2e-09) and B7-H1 (3e-08). A PSI-BLAST search based only on similarity to CD80 and CD86 identified both ICOSL sequences and B7-H1 again, but with higher significance scores (4e-15 and 3e-12 respectively). A PSI-BLAST search based on all 5 sequences (using e-7 as a significance cut-off) identified 21 proteins. -Nine of these represented members of the butyrophilin family. Butyrophilin is a cell surface protein also found in breast milk and has previously been identified as having a high similarity to the B7 family 7. -Three of the proteins corresponded to signal regulatory protein (SIRP)--1 and one to SIRP--1. Both of these are transmembrane proteins. SIRP--1 is an inhibitory receptor, expressed by splenic macrophages, which binds to the CD47 self marker on erythrocytes preventing their elimination 8. Less is known about SIRP--1, but it seems to be involved in the activation of myeloid and dendritic cells. -Four other proteins corresponding to previously cloned genes were identified: HHLA2, a human endogenous retrovirus sequence encoding a potentially secreted protein expressed in several tissues, including lymphocytes; MCAM, a melanoma adhesion protein apparently involved in tumour progression; CXADR, a surface molecule of unknown function; and VEJAM a vascular endothelial junction associated molecule, potentially involved in lymphocyte homing. In addition four novel proteins were found: IGI_M1_ctg1747_10 (2e-18), IGI_M1_ctg16974_7 (6e-10), Q9UJP1 (2e-08) and IGI_M1_ctg12704_19 (9e-08). Thus, based on some of the known proteins identified by the search, it is quite likely that some of the less characterised proteins could be involved in the co-stimulation or modulation of lymphocytes, macrophages, or other cell types. If downloading PSI-BLAST and a database is impractical, an alternative is to take the protein sequences of interest, and run them through the program BLOCKMAKER 9 to align them [http://blocks.fhcrc.org/]. The alignment can then be run through the program COBBLER 10 to obtain a consensus sequence motif. This motif can then be used to BLAST against any database of choice. We found that this works well but, since the BLAST search is based on a single sequence (albeit a consensus one), gives less significant E values than the PSI-BLAST based method. c) Finding novel molecules using structural queues: Cytokines represent an important class of immune regulators with protein folds that are particularly pliable to sequence divergence, a finding that emerges from the comparison of well-conserved three-dimensional structures that feature the faintest of chain similarities 11. In fact, striking family relationships have often emerged only after the resolution of prototype cytokine folds; for example, cementing the distant similarity between fibroblast growth factors (FGFs) and interleukin-1 (IL-1)-like molecules 12, or suggesting a link between TNF proteins and an extended family of complement C1q-like cytokines 13. These unexpected findings often broaden our view of the evolutionary emergence and biological functions of cytokines. The challenge in this genomic age is to detect novel molecules--otherwise buried in the dark recesses of sequence databases--that may have weak or unapparent ties to existing cytokine groups; we can do this best with computational techniques that are being used to sensitively annotate genome-derived sequences, and fuse knowledge of the structural templates with sensitive sequence searching and prediction routines 14. The superfamily of haemopoietic cytokines is distinguished by a unique 4-helix bundle fold that engages a special class of transmembrane receptors 11. While difficult to align by sequence similarity, the helical scaffolds of these cytokines reveal faint, subfamilydistinctive motifs when superimposed--aside from the expected register of core hydrophobic residues. Taking the best conserved 'D' helix (fourth and final in the bundle sequence) alignment of a diverse series of IL-6-like cytokine structures (comprising IL-6, GCSF, CNTF, OSM, LIF, alongside a carefully arrayed set of CT-1, IL-11 and IL-12 sequences), both weighted profiles, position-specific scoring matrices (PSSMs) and hidden Markov models (HMMs) were constructed and used to iteratively search both EST and genomic databases. This roughly 35 amino acid-long profile effectively collected all extant IL-6-type sequences, as well as a set of novel, predicted ORFs, that were then used to clone their complete gene sequences. Among these orphan cytokines is a molecule that distantly resembles CNTF and is variously called Novel Neurotrophin-1 (NNT-1) or Cardiotrophin-like Cytokine (CLC)--not surprisingly, this molecule has recently been shown to coopt the CNTF receptor complex to signal 15. Another outlier sequence has a far resemblance to the p35 subunit of IL-12, and it has recently been shown that it competes for binding to the p40 chain (creating a cytokine now labelled as IL-23), binding then to a receptor complex that includes elements of the IL-12 signalling machinery 16. In cases where no sequence similarity is detectable--but secondary structure prediction indicates, for example, a compatible register of helices and loops-- fold recognition or threading techniques are capable of teasing out a reliable alignment of a novel sequence with a helical cytokine template 17. Other forms of homology searches. As opposed to protein homology, functionally related genes may also be revealed by homology in the DNA sequence of their promoters. Gene expression profiling on DNA microarrays has revealed the co-regulation of functionally related genes. In principle this should be reflected by presence of very similar combinations of transcription factor binding sites in transcriptional control elements, such as the combined NFAT/NFkB motif in several cytokines. Correlating heritable traits with specific gene products. In practice, the genome databases are not yet at the point of listing all genes contained within a genomic interval between two markers, although it is likely that they will be within 6-12 months. As an example, consider that an autoimmune susceptibility trait has been mapped to chromosome 21 between markers D21S49 and D21S171. These flanking markers can be used to query NCBI’s Entrez Map Viewer [http://www.ncbi.nlm.nih.gov/cgi-bin/Entrez/hum_srch?chr=hum_chr.inf] which integrates human sequence and map data from a variety of sources. The types of maps include sequence, cytogenetic, genetic linkage, radiation hybrid, and YAC contig. Searching for the marker “D21S49” on chromosome “21” retrieves data from the STS sequence map only. Using the “Display settings” function on the STS map, the other flanking marker “D21S171” may now be entered. With the “STS Map” maintained as the “Master Map”, the relative positions of items on additional maps may be viewed by selecting from the various maps available (of which the “Genes_Sequence” should theoretically be the most accurate). At this stage it is also useful to increase the “Page Size” so that all markers in the chosen region are displayed. The list should now contain the genes that exist in the region between these markers. However, whilst D21S49 shows up at the top end, D2S171 is not at the bottom. Upon going back and re-searching Map Viewer for this marker it is found that is has neither been sequenced or positioned on any map available. Whilst this result seems to fly in the face of chromosome 21 being completely sequenced, it could simply be the case that the online version has not yet been annotated with all genetic markers. Two search options are now available. The first is to search the NCBI GenBank menu [http://www.ncbi.nlm.nih.gov/] as the non-redudant sequence of chromosome 21 was divided into 340-kb segments and registered in the GenBank databases under accession numbers AP001656-AP001761. Searching for “D21S171” finds the “AP001754”entry in which this marker resides (the left primer starts from basepair position 288406). An alternate search may be performed using BLAST on the human genome [http://www.ncbi.nlm.nih.gov/genome/seq/page.cgi?F=HsBlast.html&&ORG=Hs] using the left primer for D21S171 (this information is available from the STS-Based Map of the Human Genome search engine at [http://carbon.wi.mit.edu:8000/cgibin/contig/sts_info?database=release]; see also 18). This reveals one BLAST Hit aligned across the entire sequence with the reference contig “NT_002835” or “Hs21_2979”, and upon this contig resides the “AP001754” clone. The “Display settings” function on the Map Viewer may now be searched again with the flanking markers “D21S49” and “AP001754”. Maintaining the “STS Map” as the “Master Map” and selecting the “GenBank” and “Genes_Sequence” maps as additionals, brings up the region between these markers, albeit on the two different maps within which these markers reside. In the interval between the two markers, 17 candidate genes can be seen on the complete sequence map. One gene listed is a clear candidate, AIRE, which has recently been shown to carry loss-of-function mutations in a rare Mendelian syndrome, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy 19, 20. Gaps in annotation are nonetheless significant for chromosome 21, a relatively finished chromosome, as six known genes in the literature 21 between D21S49 and D21S171 (including PFKL liver-type 1-phosphofructokinase gene, positioned next to AIRE on the sequence) are not shown on Map Viewer. It is also likely that gene predictions are currently incomplete and will improve with alignment of the human and mouse sequences. By moving between different databases and using strategies outlined in a recent review 22, one can piece together a fuller picture of what is placed where on the sequence map. References 1. Baker, S.J. & Reddy, E.P. Modulation of life and death by the TNF receptor superfamily. Oncogene 17, 3261-70 (1998). 2. Screaton, G. & Xu, X.N. T cell life and death signalling via TNF-receptor family members. Current Opinion in Immunology 12, 316-22 (2000). 3. Eby, M.T., Jasmin, A., Kumar, A., Sharma, K. & Chaudhary, P.M. TAJ, a novel member of the tumor necrosis factor receptor family, activates the c-Jun N-terminal kinase pathway and mediates caspase-independent cell death. Journal of Biological Chemistry 275, 15336-42 (2000). 4. Mueller, D.L. T cells: A proliferation of costimulatory molecules. Current Biology 10, R227-30 (2000). 5. Freeman, G.J., et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. Journal of Experimental Medicine 192, 1027-1034 (2000). 6. Altschul, S.F., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25, 3389-402 (1997). 7. Henry, J., Miller, M.M. & Pontarotti, P. Structure and evolution of the extended B7 family. Immunology Today 20, 285-8 (1999). 8. Oldenborg, P.A., et al. Role of CD47 as a marker of self on red blood cells. Science 288, 2051-4 (2000). 9. Henikoff, S., Henikoff, J.G., Alford, W.J. & Pietrokovski, S. Automated construction and graphical presentation of protein blocks from unaligned sequences. Gene 163, GC17-26 (1995). 10. Henikoff, S. & Henikoff, J.G. Embedding strategies for effective use of information from multiple sequence alignments. Protein Science 6, 698-705 (1997). 11. Sprang, S.R. & Bazan, J.F. Cytokine structural taxonomy and mechanisms of receptor engagement. Curr. Opin. Struc. Biol. 3, 815-827 (1993). 12. Zhang, J.D., Cousens, L.S., Barr, P.J. & Sprang, S.R. Three-dimensional structure of human basic fibroblast growth factor, a structural homolog of interleukin 1 beta [published erratum appears in Proc Natl Acad Sci U S A 1991 Jun 15;88(12):5477]. Proceedings of the National Academy of Sciences of the United States of America 88, 3446-50 (1991). 13. Shapiro, L. & Scherer, P.E. The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Current Biology 8, 335-8 (1998). 14. Fischer, D. & Eisenberg, D. Predicting structures for genome proteins. Current Opinion in Structural Biology 9, 208-11 (1999). 15. Elson, G.C., et al. CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat Neurosci 3, 867-72 (2000). 16. Oppmann, B., Bazan, J.F. & Kastelein, R.A. IL-23. Immunity In press, (2000). 17. Madej, T., Gibrat, J.F. & Bryant, S.H. Threading a database of protein cores. Proteins 23, 356-69 (1995). 18. Dib, C., et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380, 152-4 (1996). 19. Nagamine, K., et al. Positional cloning of the APECED gene. Nature Genetics 17, 393-8 (1997). 20. The Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nature Genetics 17, 399-403 (1997). 21. Hattori, M., et al. The DNA sequence of human chromosome 21. The chromosome 21 mapping and sequencing consortium. Nature 405, 311-9 (2000). 22. Semple, C. Bases and spaces: resources on the web for accessing the draft human genome. Genome Biology 1, 1-6 (2000).