spweb chan
advertisement

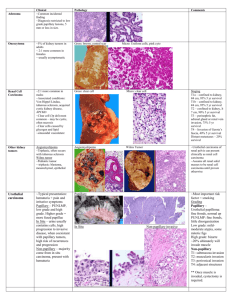
ONCOCYTOMA METASTATIC RENAL CELL CARCINOMA INVERTED UROTHELIAL PAPILLOMA SCHWANNOMA FOLLLICULAR VARIANT , PAPILLARY THYROID CARCINOMA INTRAVASCULAR LYMPHOMA Case #1: Oncocytoma Clinical History: 53-year-old male with a left kidney mass. Choose the correct diagnosis: A. B. C. Oncocytoma Renal cell carcinoma with oncocytic features Chromophobe renal cell carcinoma Histology: The tumor consists of new eosinophilic cells with small round and regular nuclei arranged in nests, cords and tubules. The background shows myxoid changes and focal fibrous scar. On high magnification, the nuclei are all uniformly round and show bland cytology without significant atypia. Small nucleoli may be seen in some of the cells. Discussion: Oncocytomas are benign renal neoplasms that are derived from intercalated cells. Grossly, the tumor appears as a mahogany, brown mass with a central scar. Histologically, the tumor is composed of eosinophilic cells with bland cytology, having small, round, regular nuclei. The tumor cells are often arranged in nests, cords or tubules in a myxoid background. Occasional degenerative atypia or pleomorphism may be seen, however, oncocytomas will not show any areas of papillary formation, necrosis or clear cells. Oncocytomas may occasionally infiltrate perirenal soft tissue, however, this does not affect the prognosis. Electron microscopy will show that the cells are packed with mitochondria, which accounts for their eosinophilic appearance. Oncocytomas may resemble chromophobe renal cell carcinomas, in that they have prominent eosinophilic cytoplasm, however, oncocytomas usually show densely eosinophilic cytoplasm throughout the entire lesion and lack the pale appearance seen in many of the cells in a chromophobe renal cell carcinoma. Chromophobe renal cell carcinomas also show perinuclear halos, and nuclear irregularity and pleomorphism, which would not be seen in oncocytomas. Chromophobe renal cell carcinomas also have distinct cell borders, which often resemble plant cells, which is a feature not seen in oncocytomas. A Hales colloidal iron stain, which stains for acidic mucin, can be helpful in cases where it is difficult to tell the two lesions apart. Chromophobe renal cell carcinomas would show intense and diffuse staining within the cytoplasm of the tumor cells, while an oncocytoma is usually negative or only shows luminal staining within some of the tubules, for Hales colloidal iron. Oncocytomas are differentiated from renal cell carcinomas with oncocytic features by the lack of cytologic atypia. Also, in renal cell carcinomas with oncocytic features, there are often areas of more conventional renal cell carcinomas showing clear cells. References: Am J Surg Pathol 1997; 21:1-12. Am J Surg Pathol 2000; 24:1247-56. CASE 2: Renal cell carcinoma, metastatic to adrenal Clinical History: 63-year-old female with adrenal mass Choose the correct diagnosis: A. B. C. D. E. Adrenal cortical adenoma Adrenal cortical carcinoma Metastatic renal cell carcinoma Pheochromocytoma Metastatic adenocarcinoma Histology: The lesion consists of a circumscribed area of clear cells growing in a nested and tubular pattern. The nuclei show some variation in size and shape, some with nucleoli. Compared to the adjacent normal adrenal tissue, the tumor cells are clear rather than vacuolated and granular. Discussion: The lesion in the adrenal raises a differential diagnosis of primary adrenal neoplasm and metastatic lesions to the adrenal gland. The lack of necrosis, nuclear anaplasia and vascular invasion, and the small size of the lesion make an adrenal cortical carcinoma unlikely. The clear cells raise the possibility of a metastasis from the kidney, which would be important to differentiate from an adrenal cortical adenoma. To unequivocally establish the diagnosis, immunohistochemical stains need to be performed. RCC, CD10, CAM 5.2, EMA are all expected to be positive in renal cell carcinoma and negative in adrenal cortical lesions, while inhibin and Melan-A should be positive in adrenal cortical lesions and negative in renal cell carcinoma. Pheochromocytomas arise from the adrenal medulla and usually shows pleomorphic cells with basophilic cytoplasm, unlike the clear cells seen in this case, neuroendocrine markers, such as chromogranin would also be positive, in contrast to the other entities in the differential. Case #3: Inverted urothelial papilloma Clinical History: 64-year-old male with hematuria. Choose the correct diagnosis: 1. Papillary urothelial neoplasm of low malignant potential with inverted growth pattern 2. Low grade papillary urothelial carcinoma with inverted growth pattern 3. High grade papillary urothelial carcinoma with inverted growth pattern 4. Inverted urothelial papilloma Histology: The tumor shows an inverted growth pattern, no papillary projections are seen. The lesion has a smooth, well circumscribed base. The cells are bland, show no mitosis and are arranged in inter-anastamosing cords. Discussion: Inverted urothelial papilloma is a rare benign tumor of the urinary bladder. Cystoscopically, it appears as solitary raised, pedunculated or, rarely, polypoid lesions with a smooth surface. Tumor size varies from small (<3 cms) to large lesions measuring up to 8 cms. Most lesions are solitary and present with hematuria. Histologically, the classic lesion shows cords and trabeculae of cells arising from a smooth surface and invaginating into the lamina propria. The periphery of the cords is composed of darker cells that are often palisade, while the central portions show maturing cells occasionally with superficial cells lining central lumenal spaces. The surrounding stroma is fibrotic and non-reactive without inflammation. Cytologic atypia is minimal to absent, although occasionally atypia of the degenerate type may be seen focally. Mitotic figures are rare, and if present, only seen at the periphery of the trabeculae. The trabeculae are orderly, of relatively uniform width, with considerable ramifications and interanastomosis. The lack of cytologic atypia, mitoses, architectural disorganization and and exophytic component rules out a papillary urothelial carcinoma with inverted growth pattern. While rare papillary urothelial neoplasms of low malignant potential may show only an inverted growth pattern, there would be some cytologic atypia and would not show the interanastamosing cords of cells with palisading nuclei seen in a benign inverted papilloma. Case #4 Schwannoma Clinical History: 52-year-old male with an intracranial mass. Choose the correct diagnosis: 1. Malignant peripheral nerve sheath tumor 2. Neurofibroma 3. Schwannoma 4. Meningioma Histology: The tumor is an encapsulated lesion, which grows eccentrically along the periphery of a nerve. The cellular tumor consists of spindle cells arranged in short bundles or interlacing fascicles and foci of nuclear palisading and whirling of cells can be seen. In some areas, the spindle cells form long sweeping fascicles. Mitotic figures are not seen. The vessels within the tumor show perivascular hyalinization. Focal small cystic areas are seen. Discussion: Classic schwannomas show a mixture of Antoni A and B areas. Antoni A areas are cellular areas composed of compact spindle cells arranged in short bundles or interlacing fascicles. In these cellular areas, there may be nuclear palisading and whirling of cells. Characteristic Verocay bodies may be seen. Antoni B areas are less orderly and less cellular areas composed of loose matrix with foci of microcystic change and inflammatory cells. This lesion lacks atypia and shows no mitoses, arguing against a malignant diagnosis. Neurofibromas may show cellular areas that resemble Antoni A areas of a schwannoma, however, unlike schwannomas, neurofibromas are not encapsulated and small nerves can usually be demonstrated within the lesion. Meningiomas can be confused with schwannomas, as cellular whirling areas can be seen in both lesions. Immunohistochemical stains can be helpful to differentiated between the two entities. Schwannomas are positive for S100 and negative for EMA, while meningiomas show the opposite staining pattern. CASE #5 FOLLICULAR VARIANT OF PAPILLARY THYROID CARCINOMA Clinical History: 33 year old woman with goiter. Choose the correct diagnosis: 1. 2. 3. 4. Papillary thyroid carcinoma Adenomatoid nodule Follicular carcinoma Follicular adenoma Histology: The tumor a well circumscribed nodule that appears distinct from the remainder of the thyroid. On low magnification, most of the tissue in the nodule appears similar to the surrounding normal thyroid. Focally, within the nodule, paler more crowded areas are seen. On higher magnification the nuclei in these crowded areas are noted to be overlapping, large, and optically clear. Nuclear grooves are also occasionally observed. Other areas with rudimentary papillary formations can be seen. Discussion: A well-circumscribed nodule is seen which contains predominantly bland follicles. Scattered throughout this nodule are areas with features including enlargement of the nuclei, clearing of the chromatin, nuclear overlapping and nuclear grooves. These features are diagnostic of papillary carcinoma and are helpful in distinguishing this nodule from follicular adenoma and graves disease, which would have more benign appearing nuclei. Since multiple areas with these classic features for papillary carcinoma were seen scattered throughout the nodule, the diagnosis of follicular variant of papillary thyroid carcinoma was made, rather than papillary microcarcinoma arising in an adenomatoid nodule. Papillary carcinoma of the thyroid is associated with the RET oncogene. The overall prognosis for patients with papillary cancer is excellent. Extra-thyroid extension into soft tissues of the neck is seen in approximately twenty five percent of patients. Involvement of cervical lymph nodes is very common and does not adversely affect the prognosis. Factors that are associated with a worse prognosis are age greater than forty years, extra thyroid extension, large tumor size, multicentricity, distant metastasis, and presence of anaplastic foci. CASE #6 intravascular lymphoma Clinical History: 66 year old man with unsteady gait Choose the correct diagnosis: 1. Metastatic carcinoma 2. Intravascular lymphomatosis 3. Brain infarction Histology: The brain parenchyma is unremarkable except for focal areas of necrosis. Large atypical cells are seen within and confined to the blood vessels within the brain parenchyma. CD 3 and CD 20 stains show that the large cells are B-cells. Discussion: Intravascular lymphomatosis (IVL) is a rare angiotrophic large cell lymphoma producing vascular occlusion of arterioles, capillaries, and venules. The majority of cases reported in the literature are case reports and small series. IVL is associated with an aggressive course with survival reported in months, and many patients are diagnosed at the time of autopsy. Antigenic phenotyping shows that these lymphomas are mostly of B cell type, and less commonly T cell or Ki-1 lymphomas. The central nervous system and skin are the two most commonly affected organs; patients usually present with progressive encephalopathy with mental status changes and focal neurological deficits and skin petechia and discoloration. Fever of unknown origin is another common presentation. Other organs can be involved, including adrenal glands, lungs, heart, spleen, liver, pancreas, genital tract, and kidneys. Bone marrow, blood, cerebrospinal fluid, and lymph nodes are typically spared. Neoplastic lymphoid cells mainly lodged in the lumina of small vessels in many organs, but infarction is most common in the CNS. Our patient presented with mental status changes, distal extremity myopathy and an elevated sedimentation rate without mass lesion in the CNS and was presumed to have a rheumatologic disorder, such as vasculitis. The histologic features of the brain biopsy are not likely to be confused with other entities. The confinement of the neoplastic cells within blood vessels, along with immunohistochemical stains proving B-cell phenotype establishes the diagnosis. The large atypical cells raise a possibility of metastatic carcinoma, however the discohesive appearance and lack of involvment of the brain parenchyma make the diagnosis unlikely. References: Mod Pathol. 1988 Nov;1(6):444-52 J Clin Pathol. 2003 Jun;56(6):468-70.