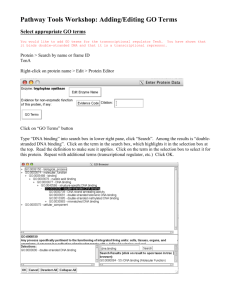
CJKinslow - Institutional Repositories
advertisement

COMMITTEE CERTIFICATION OF APPROVED VERSION The committee for Carla Jean Kinslow certifies that this is the approved version of the following dissertation: GENETIC DETERMINANTS OF NEIL2 TRANSCRIPTION Committee: ___________________________________ Sherif Z. Abdel-Rahman, Ph.D., Chair ___________________________________ Mary Treinen-Moslen, Ph.D. ___________________________________ Jonathan B. Ward, Jr., Ph.D. ___________________________________ Tapas K. Hazra, Ph.D. ___________________________________ Randa El-Zein, M.D., Ph.D. ________________________________ Dean, Graduate School GENETIC DETERMINANTS OF NEIL2 TRANSCRIPTION by Carla Jean Kinslow, B.A., M.S. Dissertation Presented to the Faculty of The University of Texas Graduate School of Biomedical Sciences at Galveston in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Approved by the Supervisory Committee Sherif Z. Abdel-Rahman, Ph.D. Mary Treinen-Moslen, Ph.D. Jonathan B. Ward, Jr., Ph.D. Tapas K. Hazra, Ph.D. Randa El-Zein, M.D., Ph.D. March, 2008 Galveston, Texas Key words: NEIL2, DNA repair, single nucleotide polymorphism © 2008, Carla Jean Kinslow Dedicated to my son, Benjamin Ellet-Kinslow and my partner, Jenifer Wagley. I could not have done this without their love and support. ACKNOWLEDGEMENTS I would like to thank my mentor, Dr. Sherif Abdel-Rahman for his patience, support, open-door, training, and respect he has given to me over the years. I would also like to thank my committee members: Drs. Jonathan Ward, Mary Moslen, Randa El-Zein, and Tapas Hazra for their support and advice. I am also grateful to Dr. Jeff Wickliffe and Dr. Marinel M. Ammenheuser for advice and critical review of the manuscript, as well as Dr. Golda Leonard for her guidance through this program. I would also like to thank all my fellow students, especially Courtney Hill for her time and patience in helping me truly understand my ‘stats’. The majority of this work was supported in part by a NIEHS Center award (ES06676), by a John Sealy Memorial Endowment (JSME) grant and by predoctoral fellowships (to C.J.K. ) from NIEHS (T32-07454). Studies were conducted with the assistance of the General Clinical Research Center at UTMB, funded by a M01 RR00073 grant from the National Center for Research Resources, NIH, USPHS. iv GENETIC DETERMINANTS OF NEIL2 TRANSCRIPTION Publication No._______________ Carla Jean Kinslow, Ph.D. The University of Texas Graduate School of Biomedical Sciences at Galveston, 2005 Supervisor: Sherif Z. Abdel-Rahman The human genome is constantly damaged by reactive oxygen species (ROS) which produce a multitude of oxidative DNA lesions that can lead to mutations, genomic instability, and ultimately, to the development of cancer and other diseases. The DNA base excision repair (BER) pathway is initiated by a DNA glycosylase and subsequently, includes several other proteins that function in a step-wise fashion to repair the mutagenic site resulting from oxidative damage. The importance of maintaining the integrity of this pathway in order to reduce disease risk has been demonstrated by many studies. NEIL2 is a novel BER glycosylase that has been associated with transcription-coupled DNA repair and has a preference for oxidative products of cytosine. Because this enzyme is thought to be involved in transcription-coupled DNA repair, it could play a substantial role in maintaining genomic stability, not only in highly proliferating cells but also in non-dividing cells, such as nerve cells, that remain transcriptionally active. Thus, alterations in the expression levels of this gene could impact DNA repair capabilities and could contribute to disease risk, especially in individuals exposed to mutagenic agents at the beginning of the study. Genetic determinants that influence NEIL2 transcription, including cis-regulatory sequences and sequence polymorphisms, were unknown. We hypothesize that cis-regulatory sequences in the 5’ upstream region of NEIL2 regulate gene transcript levels and that SNPs proximal to these sequences impact gene regulation and thus affect DNA repair efficiency, especially in individuals exposed to environmental mutagens. Consistent with these hypothesizes, we found that individual NEIL2 transcription levels varied by 63 fold amongst individuals and that sequence variations in the promoter region of the NEIL2 gene influences gene transcription levels. These sequence variations were also associated with DNA repair capacity of the cell, indicating a role for NEIL2 in regulation of global DNA repair. By characterizing the underlying mechanisms that may, in part, be responsible for variations in NEIL2 expression, we have shown that the NEIL2 promoter contains both negative and positive regulatory regions. When exposed to oxidative stress, expression from the positive regulatory region is decreased. Site directed mutagenesis of an NF-kappaB/Sp-1 site in this region abolishes this transcriptional response to oxidative stress, indicating specific cis-elements were responsible for alterations in NEIL2 in the presence of oxidative stress. Taken together, this study provides the first in vivo and in vitro descriptions of the genetic mechanisms the govern NEIL2 expression. vi TABLE OF CONTENTS PAGE ACKNOWLEDGEMENTS ............................................................................................... iv GENETIC DETERMINANTS OF NEIL2 TRANSCRIPTION........................................ vi TABLE OF CONTENTS .................................................................................................. vii LIST OF TABLES .............................................................................................................. x LIST OF FIGURES ........................................................................................................... xi LIST OF ABREVIATIONS ............................................................................................. xii CHAPTER 1: INTRODUCTION ....................................................................................... 1 General Background ........................................................................................................... 1 Oxidative DNA Damage and Disease................................................................................. 2 Base Excision Repair (BER) ............................................................................................... 4 The monofunctional glycosylases ............................................................................... 5 Bi-functional-elimination ........................................................................................ 6 Bi- functional -elimination ..................................................................................... 6 NEIL2 ................................................................................................................................. 7 Transcriptional Regulation.................................................................................................. 8 NEIL2 and transcriptional regulation .......................................................................... 9 Redox-sensitive transcription factors ........................................................................ 10 Gene Expression Studies................................................................................................... 11 The luciferase assay system ...................................................................................... 11 Single Nucleotide Polymorphisms and Gene Expression................................................. 12 Biomarkers For DNA Damage ......................................................................................... 13 Mutagen sensitivity assay ................................................................................................. 14 Tobacco-specific nitrosamines.......................................................................................... 15 Site Directed Mutagenesis ................................................................................................ 16 Objectives of the Present Study ........................................................................................ 16 CHAPTER 2: INTERINDIVIDUAL VARIABILITY IN NEIL2 GENE TRANSCRIPTION LEVELS: INFLUENCE OF SINGLE NUCLEOTIDE POLYMORPHISMS 5’ UPSTREAM OF THE CODING REGION .............................. 19 Introduction ....................................................................................................................... 19 Materials and Methods ...................................................................................................... 20 Study subjects and collection of blood samples ....................................................... 20 RNA extraction and absolute quantitation of copy numbers of NEIL2 transcript by AQ-RTPCR ............................................................................................................... 21 Determination of allelic variants in the 5’-upstream region of the NEIL2 gene ....... 22 Cytogenetic cultures for the mutagen-sensitivity assay ............................................ 23 Cell culture harvest and cytogenetic analysis for the mutagen-sensitivity assay ..... 24 Statistical analysis. .................................................................................................... 24 Results ............................................................................................................................... 25 vii Demographics of the study population. .................................................................... 25 Transcript copy numbers of NEIL2 in isolated human lymphocytes ........................ 26 Association of NEIL2 expression levels with demographic characteristics ............. 28 Relationship between NEIL2 expression and mutagen sensitivity ................................... 31 Discussion ......................................................................................................................... 32 CHAPTER 3: REGULATORY ELEMENTS RESPONSIVE TO OXIDATIVE STRESS IN THE PROMOTER REGION OF NEIL2, A HUMAN DNA GLYCOSYLASE GENE ........................................................................................................................................... 36 Introduction ....................................................................................................................... 36 Materials and methods ...................................................................................................... 37 Cell culture ................................................................................................................ 37 Mapping of the NEIL2 transcriptional start site ........................................................ 37 Cloning of the NEIL2 5’- upstream region into luciferase reporter vectors ............. 39 Sub-clone constructs of the p1200 and pSTEP3 fragments ...................................... 43 Transient transfection of MRC-5 cells with NEIL2 promoter constructs and luciferase assay ......................................................................................................... 43 Response of the p1200C and NEIL1 construct to oxidative stress ........................... 44 Statistical analysis ..................................................................................................... 44 Results ............................................................................................................................... 45 Mapping of the NEIL2 transcriptional start site and identification of cis-elements in the NEIL2 promoter. ................................................................................................. 45 Partial characterization of the NEIL2 promoter region. ............................................ 45 Response of the p1200C construct (-206 to +90 fragment) and pNEIL1-luc to oxidative stress .......................................................................................................... 49 Discussion ......................................................................................................................... 50 Chapter 4: NEIL2 gene expression is influenced by polymorphic sites as well as an NFkappaB binding motif in the promoter region................................................................... 56 Introduction ....................................................................................................................... 56 Materials and Methods ...................................................................................................... 56 In silico search for putative transcriptional binding sites ......................................... 56 Cell culture, transfection and glucose oxidase treatment.......................................... 61 Statistical analysis. .................................................................................................... 62 Results ............................................................................................................................... 62 CHAPTER 5: CONCLUSIONS ....................................................................................... 70 FUTURE STUDIES.......................................................................................................... 74 Delineation of NEIL2 regulatory motifs ................................................................... 74 Haplotype analysis of the NEIL2 promoter region ................................................... 74 Transfection of NEIL2 promoter constructs into other cell types ............................. 75 Appendix ........................................................................................................................... 76 REFERENCES ................................................................................................................. 80 VITA ................................................................................................................................. 92 Education .......................................................................................................................... 92 viii Publications ....................................................................................................................... 92 ix LIST OF TABLES PAGE Table I: Selected demographic characteristics of the study population ............................ 25 Table II: Single nucleotide polymorphism (SNP) frequency in the study population. ..... 30 Table III: Relationship between NEIL2 ss74800504 and ss74800505 polymorphisms and mutagen-induced genetic damage ..................................................................................... 32 Table IV - Primer sequences for luciferase constructs ..................................................... 42 Table V: Forward oligo primers used to introduce point mutations into luciferase plasmids. ....................................................................................................................... 57 Table VI – point mutations in pSTEP1 or p1200C constructs.......................................... 58 Table VII: Distribution of NEIL2 and XPD expression in the study population. ............ 76 Table VIII: Distribution of ethnicities, high or low NEIL2 and accumulated chromosome breaks in study population. ............................................................................................... 77 Table IX: Mean luciferase expression in NEIL2 promoter clones. ................................. 78 Table X: Mean luciferase expression in pSV-40-luciferase clones .................................. 78 Table XI: Mean luciferase expression in p1200C clones exposed to GO. ....................... 78 Table XII: Mean luciferase expression in pSTEP1 clones ............................................... 79 Table XIII: Mean luciferase expression in p1200C clones ............................................... 79 Table XIV: Mean luciferase expression in p1200C clone with mutation in the NFkB/Sp1 site. .................................................................................................................................... 79 x LIST OF FIGURES PAGE Figure 1 – Schematic model for the three BER sub-pathways in mammalian cells, modified and used with permission from Weiderhold et al. (2004). .................................. 5 Figure 2 – Distribution of NEIL2 expression in the study population (n=129). Individuals having higher than 6900 copies/g of total RNA were designated as “high expressers” (12%) and those having less than 4150 copies/g total RNA were designated as “low expressers” (88%). ............................................................................................... 27 Figure 3 (a) – Distribution of NEIL2 expression stratified by ethnicity (grey bars). *P<0.05 for comparison between White non-Hispanics and other ethnic groups combined, indicate a significantly higher mean NEIL2 transcript copy numbers in White non-Hispanic subjects. Values are expressed in mean ± SE. ........................................... 29 Figure 3 (b) Distribution of NEIL2 transcript copy numbers amongst the different ethnic groups. ............................................................................................................................... 29 Figure 4: RACE methodology .......................................................................................... 39 Figure 5: Schematic representation of the NEIL2 gene and the relative locations of the promoter fragments isolated, cloned, and sub-cloned into luciferase reporter vectors. ... 40 Figure 6: The DNA sequence, encompassing the pSTEP1 fragment, that includes the NEIL2 gene sequence flanking 5’ upstream and 3’ downstream of the identified transcriptional start site (+1). ............................................................................................ 42 Figure 7: A, Luciferase constructs containing DNA fragments of the 5'-regulatory region that were cloned upstream of the luciferase-coding sequence; B, Promoter activity ....... 46 Figure 8: A, Luciferase constructs containing the pSTEP3 DNA fragment inserted 5’ of a SV-40 promoter; B, Promoter activity relative to a pSV-40 control vector is shown in relative light units (RLUs) per mL, normalized to total protein in the sample. ................ 47 Figure 9: A, Luciferase sub-clone constructs containing fragments (A, B, and C) of the p1200 DNA fragment inserted upstream of luc-coding sequence; B, Promoter activity . 49 Figure 10: Luciferase reporter activity in cells transfected with the p1200C and pNEIL1 constructs and then treated with glucose oxidase (100 ng/mL) for 1, 6, and 12 hrs......... 50 Figure 11: The DNA sequence, encompassing the pSTEP1 fragment, that includes the NEIL2 gene sequence flanking 5’ upstream and 3’ downstream of the identified transcriptional start site (+1). ............................................................................................ 61 Figure 12: Promoter activity from pSTEP1 constructs containing site-directed mutations ........................................................................................................................................... 63 Figure 13: Promoter activity from p1200C constructs containing site directed mutations ........................................................................................................................................... 64 Figure 14 Promoter activity for p1200C wild type and p1200C + mutation in NFkappaB site (104 GtoC) with and without 1 hr GO treatments ...................................................... 65 xi LIST OF ABREVIATIONS DNA; deoxyribonucleic acid RNA; ribonucleic acid ROS; reactive oxygen species APE; apurinic/apyrimidinic endonuclease NF-kappaB; nuclear factor kappa B OGG1; 8-oxoguanine DNA glycosylase NTH1; endonuclease III homolog SNP; single nucleotide polymorphism LUC; luciferase gene RLU; relative light units CAT; chloramphenicol acetyl transferase CA; chromosome aberration GO; glucose oxidase NNK; 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone PHA; phytohemagglutinin PBL; peripheral blood lymphocyte PCR; polymerase chain reaction AQ-RTPCR; absolute quantitative reverse transcription PCR xii CHAPTER 1: INTRODUCTION General Background Just prior to initiation of the Human Genome Project, James D. Watson stated, “When finally interpreted, the implications of the genetic messages encoded within our DNA molecules will provide the ultimate answers to the chemical underpinnings of human existence” (Watson, 1990, p.44). This full sequence of the human genome was sequenced in 2000 by a private group of researchers headed by J. Craig Venter and very shortly thereafter by a NIH-backed group led by Fransis Collins (Venter et al., 2001; Lander et al., 2001). This sequence was thought to somehow, perhaps magically, reveal the answers to all of our unanswered biological questions (Venter et al., 2001). One of the most obvious and critical question to answer was, “What makes some people acquire a disease and others not?” Unfortunately the sequence, itself, did not give researchers the answer to this broad question but is now allowing molecular epidemiologists to ask more specific questions, such as“What are the environmental and genetic factors that determine whether a specific group of people acquire a disease, when other groups do not?” Because the entire genome is now available for analysis, researchers can identify subtle, inheritable differences, such as single nucleotide polymorphisms (SNPs, single base changes occurring in greater than 1% of a population) amongst diseased and nondiseased groups. After establishing associations between SNPs and to disease risk, scientists are now able to better identify the common genetic and environmental factors amongst diseased and non-diseased populations. This is the first step in understanding the dual contribution of biological and environmental factors to disease risk. In this way, studies showed that individual disease susceptibility does not depend solely on genetic factors, but environmental factors, including diet, exercise, exposure to toxins, and levels of stress are important as well. 1 Oxidative stress is one of the most common and the most detrimental environmental factors linked to disease risk (reviewed by Karihtala and Soini, 2007; Flora, 2007). Humans are constantly exposed to oxidative stress from both endogenous and exogenous agents that generate free radicals and reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), superoxide (O•2-) and the highly reactive hydroxyl (•OH) radicals (Pages and Fuchs, 2002; Gros et al., 2002). Fortunately, human cells have inherent mechanisms available to detoxify ROS. These mechanisms include simple defenses, such as those provided by the antioxidants vitamin C and vitamin E, which react with, and lessen the reactive nature of the ROS (Evans et al., 2004). Other mechanisms involve the actions of detoxifying enzymes, such as superoxide dismutase, to intercept and neutralize ROS (Evans et al., 2004). The protective effect of the cell is directly associated with the capability of the cell to remove these damaging molecules; however, excessive oxidative stress can overwhelm the protective machinery (Hazra and Roy, 2007; Gros et al., 2002). Excessive amounts of ROS, if not detoxified can cause severe cellular injury or even cell death (Gros et al., 2002). In the presence of excessive oxidative stress, ROS attack the DNA, frequently producing pro-mutagenic DNA lesions (Weiderhold et al., 2004). These lesions have been linked to several disease processes, including aging and cancer (Gros et al., 2002). Oxidative DNA Damage and Disease Mitochondria are the primary intracellular sources of ROS, which are generated from re-dox reactions during normal cellular metabolism (Evans et al., 2004). ROS are produced from as much as 5% of the consumed O2 during the process of energy production in the mitochondria (Hazra and Roy 2007). External sources of ROS include UV radiation and tobacco smoke (Weston and Harris, 2003; Karihtala et al., 2007) leading to deregulation and damage. Such sources can increase cellular oxidative stress, creating an environment conducive to depleting the cellular antioxidant defenses (Weston and Harris, 2003; Karihtala et al., 2007). In the cytoplasm, ROS can readily enter the 2 nucleus and attack DNA (Pages and Fuchs, 2002). Oxygen radicals can attack various sites throughout the DNA double-helix, including each of the four bases (Kuraoka et al., 2007). Oxidative attack on DNA bases can result in at least 80 different forms of DNA base modifications (Kuraoka et al., 2007). The frequency, persistence in reactivity, and the large number of oxidized DNA bases that are produced from ROS can lead to genomic instability. This instability has been associated with the development of various diseases (Karihtala et al., 2007; Evans et al., 2004). Cancer is one of the most thoroughly characterized diseases associated with the effects of oxidative stress (Evans et al., 2004; Flora, 2007). Several oxidized DNA bases have been identified as appropriate biomarkers for cancer and certain other diseases, indicating that oxidative DNA damage has a direct impact on disease risk (Evans et al., 2004). For example, 8-hydroxydeoxyguanosine (8-OH-dG) is a wellcharacterized mutagenic lesion, in addition to being a known biomarker for oxidative stress (Evans et al., 2004). This damaging lesion has been found in liver cancer, skin cancer, and lung cancer (Jungst et al., 2004; Nishigori, 2004; Paz-Elizur et al., 2005). There is also an association between inflammation and elevated levels of 8-OH-dG. Elevated levels of this lesion were reported in lymphocytes of patients with rheumatoid arthritis and systemic lupus erythematosus (Evans et al., 2004). The results from several studies have also suggested that oxidized lesions are associated with neurological diseases, (including Alzheimer’s, Huntington’s, and Parkinson’s disease) as well as cardiovascular disease and aging (reviewed by Evans et al., 2004; Flora, 2007). The removal of oxidized DNA bases is essential in order to prevent diseases that are associated with these lesions (Fortini et al., 2003). The base excision repair (BER) pathway recognizes and removes oxidized base lesions; however, inefficiencies in this enzymatic cascade can result in an accumulation of oxidized bases in DNA, leading to genomic instability. Epidemiological studies have shown that polymorphisms in several BER enzymes, including 8-oxoguanine DNA glycosylase (OGG1) and apurinic/apyrimidinic endonuclease are associated with cancer (Zidnoddiny et al., 2005; Paz-Elizur et al., 2005; Kiyohara et al., 2006; Li et al., 2007). Specifically, certain non3 synonomous SNPs, that alter structure in the coded protein, in the BER enzyme OGG1 have been associated with reduced activity of this enzyme in humans. This reduced activity was associated with elevated cancer risk (Fortini et al., 2003). Also, an increased risk of lung cancer was reported in aging Ogg1 knock-out mice (Sakumi et al., 2003). Base Excision Repair (BER) The role of the BER pathway is in preserving DNA integrity and cellular stability (Krokan et al., 2000). The first step (Step 1) of the BER pathway is the removal of the damaged base by a DNA glycosylase. This is followed by abasic site priming (Step 2), gap filling, and ligation (Figure 1, Step 3) of the DNA (Evans et al., 2004). In humans, there are two groups of DNA glycosylases that initiate the first step. They are defined by their method of base removal, which is either as monofuctional or bi-functional glycosylase (Weiderhold et al., 2004; Figure 1). The importance of this repair process has prompted researchers to investigate imbalance in expression levels of BER enzymes. For example, Fortini et al. (2003) found that a burst in activity of alkylpurine-DNA-glycosylase (MPG, a BER enzyme that removes N-alkylpurines) creates partially repaired sites. Furthermore, up-regulation of this gene has been seen in breast cancers (Fortini et al., 2003), suggesting that the imbalance of the enzymes in the BER pathway can potentially lead to un-repaired DNA damage in cells that, in turn, can lead to increased disease risk. 4 Step 1 Step 2 Step 3 Each sub-pathway is characterized by its respective glycosylases OGG1 and NTH1, monofunctional glycosylases (M), or NEIL1 and NEIL2. Figure 1 – Schematic model for the three BER sub-pathways in mammalian cells, modified and used with permission from Weiderhold et al. (2004). The monofunctional glycosylases Some of the earliest studies on BER suggested a streamlined theory with regard to the mechanism of oxidized lesion repair, where a specific glycosylase would remove only one type of oxidized lesion (Izumi et al., 2003). The types of glycosylases in this pathway include uracil-DNA glycosylases, which remove uracils from the DNA strand and 3-methyl-DNA glycosylases, which remove alkylated DNA bases (Korolev, 2005). Today, it is known that these enzymes are monofunctional glycosylases. These glycosylaes use an activated water molecule to catalyze a hydrolytic reaction across the 5 glycosydic bond (McCullough et al., 1999). This removes the damage base and results in the formation of an abasic site (McCullough et al., 1999; Izumi et al., 2003; Weiderhold et al., 2004). After this step, the 3’ terminus of the AP site must then be modified to 3’OH prior to complete strand repair. This is accomplished by the action of the APendonuclase (APE) enzyme, followed by final repair of the strand by DNA polymerase and DNA ligase (Evans et al., 2004). Bi-functional-elimination -elimination of oxidized bases is initiated by either the OGG1 or endonuclease III homolog (NTH1) glycosylases (Weiderhold, 2004). These are bi-functional glycosylases that belong to the endonuclease III (Nth) superfamily and have glycosylase as well as intrinsic lyase activity that can modify the abasic site to an --unsaturated aldehyde after -elimination (Weiderhold et al., 2004). The --unsaturated aldehyde is further modified by addition of a 3’OH by APE (Weiderhold et al, 2004). Although both of these enzymes remove the damaged base by using the same process, the OGG1 enzyme specifically removes the oxidized guanine, 8-oxo-dG, whereas NTH1 removes oxidized pyrimidines (Weiderhold et al., 2004). Bi- functional -elimination In 2002, a new sub-class of DNA glycosylases was characterized in which both -elimination and AP lyase activity were used to remove and modify the damaged base, as shown above in Figure 1 (Wiederhold et al., 2004, Hazra, et al., 2002). Initial characterization of this novel class of enzymes revealed that these proteins demonstrate enzymatic activity similar to that of the E. coli enzymes, Nei and Fpg, thus they were named Nei-like (NEIL) (Bandaru et al., 2002; Hazra et al., 2002a, 2002b; Morland et al., 2002; and Takao et al., 2002a). The NEILs recognize and remove the damaged base, leaving an abasic site and a break in the DNA strand (Weiderhold et al., 2004). These enzymes then catalyze the elimination at the AP site, which leaves a 3’ phosphate in the strand break (Weiderhold et al., 2004). 6 This site is further modified by polynucleotide kinase (PNK) (Weiderhold et al., 2004). An inorganic phosphate is removed from the 5’ side of the AP site to generate a typical 3’OH, which can be used in a base addition reaction. Repair of the site is completed by DNA polymerase and DNA ligase (Weiderhold et al., 2004). Further characterization of the NEILs has shown that they excise 5-hydroxyuracil (5-OHU) and thymine glycol, as well as 8-oxo-dG (Wiederhold et al., 2004). To date, three NEIL proteins have been described, NEIL1, NEIL2, and NEIL3 (Hazra et al., 2002b; Morland et al., 2002). Of these three, NEIL1 and NEIL2 have been most characterized. These two proteins have a preference for excising the highly mutagenic 5-hydroxyuracil from a DNA bubble and from double strand structures, suggesting that they are likely associated with repair during transcription (Hazra et al., 2002). Specifically, NEIL2 has an affinity for oxidized cytosine, which is one of the most mutagenic and prevalent of the oxidized lesions (Dou et al., 2003; Hazra et al., 2002a; Hazra et al., 2002b; Dou et al., 2003). The results from many studies indicate that NEIL2 holds an influential role in DNA repair. Thus, an alteration in the amount or the activity of this enzyme could have profound and long-term impacts on overall genetic stability (Dou et al., 2003; Hazra et al., 2002a; Hazra et al., 2002b; Dou et al., 2003). Yet, due to its novelty, the factors that could alter the levels of NEIL2 protein expression and the down-stream affects of such alterations have yet to be elucidated. The current project investigates these genetic factors and how they could be related to disease risk. NEIL2 NEIL2 has quite a unique enzymatic nature. In addition to its substrate preference, it has a high affinity for excising damage (such as the DNA lesions spiroiminodihydantoin and guanidinohydantoin) caused by carcinogenic metals, whereas other BER enzymes, such as OGG1 and NTH1, have almost no affinity for these lesions (Hailer et al., 2004). Furthermore, there is growing evidence that the NEIL2 enzyme has a distinct affinity for repairing lesions in DNA bubble structures (Dou et al., 2003). This 7 is exceptional and is in contrast to other BER proteins, such as OGG1 and NTH1 that only act on duplex DNA (Dou et al., 2003). The ability of NEIL2 to repair lesions in DNA bubble structures suggests that alteration in expression and/or function of NEIL2 could impair the efficiency of repairing oxidative damage during transcription. This also suggests that NEIL2 could play a particularly important role in DNA repair of highly proliferating tissues, a hallmark process in cancer development. Consistent with this suggestion, in a recent study, Broderick et al. (2006) reported an association between a silent mutation in the NEIL2 gene and increase risk for gastric cancer. This study suggests a genetic link between NEIL2 and disease risk; however, the mechanisms underlying this association have not been characterized. Furthermore, to our knowledge, the genetic factors regulating the expression of NEIL2 have not been investigated. For example, there are several reported SNPs in the 5’-upstream region of NEIL2; however, their effects on the transcription of the gene, and/or their associations with disease risk, are still unknown (National Center for Biotechnology Information, dbSNP). A growing number of studies have characterized the NEIL2 protein; yet, few studies have delved into the genetics of this gene. Thus, there are several gaps in knowledge regarding the understanding of how the transcription of NEIL2 is regulated, how polymorphisms in the promoter of this gene impact its expression in vivo, and how changes in its expression could be associated with disease risk. These are gaps in knowledge that are addressed in the current project. Transcriptional Regulation Transcriptional regulation is the most common form of gene control in eukaryotes (Alberts et al., 2002). Usually, transcription is initiated at or near a specific location in the template DNA (the start site) (Alberts et al., 2002). Transcription is regulated by trans-acting proteins (transcription factors) that bind to specific DNA sequence motifs (cis-acting sequences). Cis-acting sequences can be located in various regions of the gene, commonly the region 5’-upstream of the transcriptional start site (Hasselbach et al., 8 2005). The transcriptional regulatory proteins that bind to these sites play a role in recruiting the transcriptional machinery, such as RNA polymerase, to the start site (Hasselbach et al., 2005; Gazzoli and Kolodner, 2003). Control of gene expression is accomplished through a complex interplay between the position of these DNA sequences and the transcriptional activating proteins that bind to them. In this way, both protein and DNA sequence are involved in the regulation of gene expression. The DNA binding motifs not only site for the bind of positive-regulating proteins, to initiate gene expression, but also for the binding of repressor proteins to silence transcription (Ogbourne and Antalis, 1998). If there is a sequence change (such as a polymorphism, for example) in or near one of these DNA motifs, the respective transcriptional protein element might not recognize it and bind to it, resulting in an alteration in the levels of transcription. In fact, several epidemiological studies that have associated promoter polymorphisms in several genes to increased disease risk (Taioli et al., 2007; Pereira et al., 2007; Hoshi et al., 2007; Wang et al., 2008). On the other hand, changes in the sequence of these motifs could result in the enhancement of transcription. This would not always be a favorable response since in a pathway such as BER that depends on precise interplay of protein function, an excess of one enzyme in the enzymatic cascade could result in dysfunctional DNA repair. In this way, integrity of the promoter sequence must be retained in order to maintain proper equilibrium of gene transcription, thus protein balance. NEIL2 and transcriptional regulation During the initial characterization of NEIL2, Hazra et al. (2000 and 2002a) showed that NEIL2 was differentially expressed in multiple human tissues (Hazra et al., 2002a; Hazra, et al., 2002b). Although all tissues of an individual have the same gene sequence, differential gene expression patterns between tissues can be caused by several factors, including: (1) localized exposure to external sources of cellular stress (such as tobacco smoke in the lungs), (2) localized cellular damage (such as inflammation at a site 9 of injury), and (3) normal metabolism of that tissue (such as the high rate of metabolism of muscle cells that creates excess ROS leakage from the mitochondria) (Niess and Simon, 2007; Ji, 2007). Hazra et al. (2002a) showed that NEIL2 expression was highest in muscle tissue, suggesting that it may be over expressed in a highly metabolic tissue, which creates substantial intercellular ROS (Niess and Simon, 2007; Hazra et al., 2002a; Ji, 2007). Thus, ROS may play a role in up-regulation of the NEIL2 gene. Furthermore, the differential expression of NEIL2 suggests that it’s level of transcript is not static, thus it could be affected by external or localized factors. This may be related to regulatory regions in the NEIL2 gene that may respond to different cellular conditions found in different tissues. As such, the patterns of protein expression may vary amongst individuals, and amongst other tissues, if there are SNPs in cis-acting motifs found in the 5’-upstream region regulating level of gene expression. Such a sequence change could impact the binding affinity of the trans-acting proteins, resulting in a differential expression of the transcript (Wang et al., 2006). Such alterations were exemplified in a study of the ABCB1 gene, where 5’-upstream haplotype (a set of closely linked polymorphisms) appears to be associated with a decrease in transcription levels in HeLa (epithelial) cells, but with an increase in levels in HEK293 (kidney) cells (Wang et al., 2006). The transcript of NEIL2 could be regulated in a similar manner by SNPs in the in the promoter region of the NEIL2 gene. Redox-sensitive transcription factors There are several groups of transcriptional proteins that are activated during increased oxidative stress, but the most characterized include nuclear factor kappa-B (NF-kB), activator protein-1 (AP-1), and the p53 family of proteins (Evans et al., 2004; Rahman, 2003; Marthy-Hartert et al., 2003). Of these, NF-kB and AP-1 are associated with the expression of various DNA-repair-related associated proteins such as NEIL1, ERCC1, MGMT, and APE (Li et al., 1999; Das et al., 2005; Boldogh et al., 1998, Xu et al., 1997). Several studies have shown that ROS induces DNA repair by mechanisms 10 mediated by transcription factors (Evans et al., 2004). Das et al. (2005) has shown that NEIL1 gene expression is induced by acute exposure to glucose oxidase (a strong ROS generator), suggesting that the genes in this sub-pathway respond to oxidative stress (Das et al., 2005). Excessive oxidative DNA damage has been shown to block transcription of the BER glycosylase, OGG1 (Hailer-Morrison et al., 2003). However, how such a situation affects the NEIL2 gene is unknown. Gene Expression Studies The luciferase assay system The firefly luciferase protein was initially purified in 1978, and functional characterization of this very stable bio-luminescent protein soon followed (DeLuca and McElroy, 1978). A decade later, the gene was cloned from a gt11 Photinus pyralis cDNA library by De Wit et al. (1987). This began a new and dynamic direction for mammalian expression studies with the development of the luciferase expression system (De Wit et al., 1987). This expression system overcame several short-comings of other expression systems such as bacterial -galactosidase (-gal) and chloramphenicol acetyl transferase (CAT). The advantages of luciferase over these other systems include easier one-step quantification of promoter activity, increased protein stability, higher sensitivity, lower requirements of initial material (1/10 the cells used in the CAT system), and a shorter time from transfection to quantitative measurement of promoter activity (Williams et al., 1989). Since then, the design and application of the firefly luciferase gene in genetic reporter systems have contributed greatly to the understanding of gene expression and regulation (Gould and Subramani, 1988). For use in most gene expression studies, the luciferase reporter gene is joined to a promoter sequence in an expression plasmid. The newly constructed plasmid is then transfected into living cells and then the cells are assayed for the presence of the reporter gene by measuring the enzymatic activity of the reporter protein. This technique allows for gene expression to be assayed within a human cell under a host of experimental 11 conditions. The luciferase reporter gene is very close to an ideal reporter gene for our studies because it is not endogenously expressed in mammalian cells, is a sensitive assay that is quantitative, rapid, easy, reproducible and safe (Gould and Subramani, 1988). In the current studies, this reporter assay system was utilized to characterize the NEIL2 promoter region. Briefly, fragments of genomic DNA corresponding to the putative NEIL2 promoter region were cloned in front of the luciferase gene and then evaluated by promoter-driven expression of each fragment. The design of this assay system allowed for the evaluation of expression from various plasmids that contained engineered polymorphisms as well as the ability to evaluate NEIL2 promoter-driven expression during conditions of cellular oxidative stress. Single Nucleotide Polymorphisms and Gene Expression Single nucleotide polymorphisms (SNPs) are single base differences in the DNA strand that occur at a frequency greater than 1% in the population (National Center for Biotechnology Information, online SNP database; Weiss, 1998). A SNP can occur in several places throughout a gene. Many of the SNPs that have been reported occur in the non-coding regions. For several genes, SNPs in promoter sequences result in alterations in gene expression (Wang et al., 2006; Kim et al., 2006; Southam et al., 2007;Faniello et al., 2006). In a number of studies, significant increases or decreases in gene expression in the presence of promoter SNPs have been described (Wang et al., 2006; Kim et al., 2006; Southam et al., 2007; Faniello et al., 2006). Because promoter regions contain both positive and negative regulatory sequences, promoter SNPs have been associated with increased as well as decreased expression (Wang et al., 2006; Kim et al., 2006; Southam et al., 2007;Fanielle et al., 2006). A single base change could cause an increase in expression by creating a new protein binding site or by enhancing the binding of a transcriptional protein to a DNA sequence. Alternatively, it may abolish the binding of a positive regulatory protein or even create a site for the binding of a negative regulatory protein, resulting in decreased gene expression. 12 Thus, a change in expression is, presumably, due to an alteration in factor binding due to modification of the DNA sequence. Precedents are known for several DNA repair genes, including MSH6 and Rad51 (Gazzoli and Kolodner, 2003). For example, there is a polymorphism that disrupts a Sp-1 binding site of the MSH6 repair gene promoter and results in a 50% drop in expression (Gazzoli and Kolodner, 2003). Several researchers have reported a possible link between the presence of 5’-upstream SNPs in DNA repair genes and progression of cancers such as gastroesophageal, breast, and melanoma (Hasselbach et al., 2005, Tan et al., 2005, and Egyhazi, 2002). Furthermore, there is a growing body of epidemiologic data associating promoter polymorphisms to disease risk (Villard, 2004; Taioli et al., 2007; Pereira et al., 2007; Hoshi et al., 2007; Wang et al., 2008). In patients with the disease hemophilia B Leyden, a mutation at the -20 site (20 base pair 5’ of the transcriptional start site) disrupts the binding of hepatocyte nuclear factor 4 (NFN4) (Villard, 2004). This prevents the transcription of plasma factor IX. Thus, these individuals do not have some of the essential clotting factors (Vallard, 2004). Other examples of promoter polymorphisms that are associated with disease include a SNP in the tumor necrosis factor alpha (TNF promoter that result in an increased risk of death from cerebral malaria and a SNP producing a promoter variant of the CCR5 gene (which encodes for a cell surface chemokine) that is associated with the progression of AIDS (Vallard, 2004), however the exact mechanisms are not known. Biomarkers For DNA Damage Biomarkers are measurable characteristics that can be used as indicators of biological response (Dalle-Done et al., 2006). Biomarkers often are selected as a means to identify subtle cellular changes that occur early in a well-characterized disease pathway (Dalle-Done et al., 2006). Important characteristics of biomarkers are that they can be objectively evaluated, have a high specificity, and are not too invasive for use in 13 an in vivo investigation (Dalle-Done et al., 2006). In the present study, chromosome aberrations were used as a biomarker of early biological effect. Chromosome aberrations (CA) are distinct changes in chromosome structure, such as strand breaks and rearrangements (Bonassi, 1995). They are indications of dynamic genotoxicity and are the only biomarker that has been shown to be a predictor of cancer susceptibility (Hagmar 1994; Bonassai, 1995; Bonassai and Hagmar, 1998). Several studies have shown that environmental sources of reactive oxygen species, such as tobacco smoking and ionizing radiation, are associated with genomic instability and chromosome aberrations (El-Zein et al., 2000; Jones et al., 2007; Williams et al., 2005; Luo et al., 2004). Mutagen sensitivity assay The mutagen-sensitivity assay is a well-established technique that has been used to elvaluate an individual’s response to the mutagenic effects of using cells from that individual, (reviewed by Wu et al., 2005). This assay, developed by T.C.Hsu in 1989, is based on isolating cells, such as peripherial blood lymphocytes (PBL’s), from an individual and then exposing these cells to genotypic compounds. After exposure, cells are evaluated for genotoxic effects, such as chromosome aberrations or sister chromatid exchanges. This assay reflects an individual’s sensitivity to the mutagen used in the assay, as well as the DNA repair capacity of that individual. This assay has been used in several studies to show that PBL’s from subjects with tobacco-related cancer have higher mutagen-sensitivity when compared to the cells of non-cancer control subjects (Wu et a;., 2006; Shen et al., 2003). Furthermore, a study by Blasiack et al., (2004) reported that PBL’s of breast cancer patients have reduced DNA repair kinetics and increased DNA damage following exposure to hydrogen peroxide (a strong ROS generator) and doxorubicin. Thus, we chose to use this assay to test the PBL’s from our population for genotoxic effects of the tobacco-specific mutagen, using the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) as our mutagen. 14 Tobacco-specific nitrosamines Cigarette smoke contains over 60 carcinogens (Hoffmannet al., 2001). The tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is a strong pulmonary carcinogen and a potent inducer of lung adenocarcinoma, now the leading lung cancer subtype in the United States (Thun et al., 1997). NNK induces lung cancer independent of route of administration, in both susceptible and resistant strains of mice (Hecht and Hoffmann, 1989). Studies on the metabolism of NNK have shown that it induces cross-links in DNA; interacts with DNA, forming different types of adducts; and increases the frequency of chromosome aberrations (Weitberg and Corvese, 1993, Berwick and Vineis, 2000). DNA adducts are generated by NNK through the methylation or the pyridyloxobutylation pathway. Another study by Chuang and Hu (2006) has shown that preincubation of stimulated white blood cells with NNK, followed by UV irradiation synergistically increased DNA damage, lipid peroxidation, and the level of intracellular ROS. Thus, exposure to NNK can cause a diverse genotoxic response, resulting in various types of DNA adducts and lesions. Many of these adducts have been detected in cells and tissues susceptible to NNK carcinogenesis in rodents and humans (Hecht, 1999). Therefore, maintaining the integrity of the cellular DNA repair process is essential in protecting the cell from genomic instability that results in carcinogenesis. The repair kinetics for NNK-induced genetic damage has not been clearly elucidated but may involve several DNA-repair pathways, including base excision and nucleotide excision repair pathways (Cloutier et al., 2001). In the current study, we used NNK to induce genotoxic effects in th ePBL’s of our population. 15 Site Directed Mutagenesis Site-directed mutagenesis is a molecular technique that enables the researcher to make a deliberate, one base pair change in a sequence, while leaving the remaining sequence unchanged. This technique was initially developed in 1974 by Flavell et al. and since then, has been used in thousands of experiments (NCBI, 2008), thus it has become one of the essential molecular techniques used today. This technique was used in the present study to introduce single base changes in the NEIL2 promoter luciferase reporter plasmids at the locations of previously reported SNPs. In this way, the effects of a single base change on the relative activity of the promoter were characterized, independent of other SNPs within the sequence. The effects of combinations of these SNPs engineered into these constructs were also be characterized. Objectives of the Present Study The objective of the current study was to address a number of gaps in the understanding of NEIL2 transcriptional regulation as well as its potential role in disease risk. In order to better understand the effect of variation in NEIL2 gene transcription on levels of genetic damage in humans, an absolute quantitative reverse transcription PCR (Q-RT-PCR) analysis of NEIL2 gene expression within a population of smokers and nonsmokers was conducted. The hypothesis is that NEIL2 transcription varies considerably among individuals, and that this variability is influenced by genetic, as well as, by environmental factors. A secondary hypothesis is that reduction in transcription is associated with increased genetic damage in subjects exposed to environmental mutagens, such as those found in tobacco smoke. In order to characterize the underlying genetic mechanisms that could, in part, be responsible for variation in NEIL2 expression, an in silico analysis was performed using the web-based TESS (Transcriptional Element Search System). This web-based software program was used to search the sequence proximal to the NEIL2 start site and is designed to identify cis-regulatory sequences corresponding to various transcription factors. The results of this analysis will be used to generate a putative map containing the locations of 16 the cis-regulatory sequences in the NEIL2 5’-upstream region. This map was used to guide the generation of luciferase reporter vectors encompassing regions of the NEIL2 upstream region that are rich in cis-acting motifs. These reporter plasmids were tested in an in vitro luciferase reporter assay system and the level of reporter gene expression was related to the impact of each sequence on gene expression. The hypothesis was that NEIL2 transcript level is regulated by the sequence located 5′-upstream of the transcriptional start site. The premise was that the cis-acting regulatory motifs located within this 5’ upstream region that govern this transcriptional regulation can be identified by applying site directed mutagenesis to alter the putative regulatory sequence in the reporter plasmid. The level of promoter-driven expression from these mutated plasmids was then addressed in vitro and related to that of the wild-type plasmids. In order to integrate the information generated from the studies above, an a priori exploratory genotype-phenotype analysis was performed using lymphocyte samples from a study population composed of healthy individuals. A sub-population of individuals was selected for sampling and a fragment in the NEIL2 promoter region was sequenced. The frequencies of SNPs was evaluated for associations with numbers of NEIL2 gene transcripts and with mutagen sensitivity. Results from these analyses drove further investigations towards a mechanistic understanding of how the associated promoter SNPs in NEIL2 could be associated with these factors. The mechanistic relationships of relevant SNPs was studied in vitro using site directed mutagenesis and the luciferase expression system. NEIL2 reporter plasmids that were created in the studies described above, were used as templates for site-directed mutagenesis to introduce SNPs into the promoter fragment of the plasmids. The effect of each of the SNPs on luciferase-driven expression was tested in vitro. These experiments will provide a mechanistic explanation for associations with either NEIL2 expression or mutagen-induced genetic damage observed in vivo. Collectively, results from these experiments will provide a linear mechanistic link between levels of NEIL2 expression variability in vivo and the genetic factors that are responsible for such differences in NEIL2 expression. From a scientific perspective, the 17 goal was to provide a better understanding the factors regulating this important gene. From a public health perspective, the goal was to help identify sub-groups of the population that could be at a higher risk of disease from external environmental factors that increase levels of RIS, such as those associated with tobacco smoking. 18 CHAPTER 2: INTERINDIVIDUAL VARIABILITY IN NEIL2 GENE TRANSCRIPTION LEVELS: INFLUENCE OF SINGLE NUCLEOTIDE POLYMORPHISMS 5’ UPSTREAM OF THE CODING REGION Introduction Several SNPs have been reported in genomic regions located 5’ of the coding region of the NEIL2 gene (NCBI on line data base, 2008). These SNPs, designated as regulatory SNPs (rSNPs), could affect transcriptional regulation (Mottagui-Tabar et al., 2005, Cheung et al., 2005; Mohrenweiser, 2007). The regions contain key transcriptional regulatory elements (cis-elements) that encode short (less than 25 bp) sequences that serve as binding motifs for important transcriptional proteins (Mottagui-Tabar et al., 2005, Cheung et al., 2005; Mohrenweiser, 2007). Regulatory SNPs have been reported in various DNA repair genes, and studies have clearly indicated that these rSNPs could seriously impact the regulation of gene transcription (Mohrenweiser, 2007; Hasselbach et al., 2005; Tan et al., 2005), and accordingly, could affect disease risk (Wang et al., 2006a; Wang et al., 2006b; Wang et al., 2006c; Lu et al., 2007; Hu et al., 2007). Nevertheless, rSNPs in the 5’ upstream regions of DNA repair genes have not been thoroughly studied with respect to disease risk compared to SNPs in the coding regions. The effects of SNPs in the 5’ upstream region of the NEIL2 gene on gene transcription and/or their associations with disease risk, either individually or as a part of common haplotypes, are still unknown. In the current study, we test the hypothesis that NEIL2 transcription is influenced, at least in part, by the inheritance of SNPs in the 5’region of the gene. In addition, we also test the hypothesis that perturbation of NEIL2 expression is associated with increased genetic damage in individuals exposed to genotoxic environmental agents such as those found in tobacco smoke. In the current studies, the mutagen-sensitivity assay (Hsu et al., 1989) was used, with the tobaccospecific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) as the test 19 mutagen. This assay measures the DNA repair capacity of cells exposed to this mutagen. NNK stimulates the formation of ROS (Kim and Wells, 1996; Rioux and Castonguay, 2000; Wells et al., 1997; Chuang and Hu, 2006) that form oxidative DNA lesions, as well as strand breaks and chromosome aberrations (Chuang and Hu, 2006; Weitberg and Corvese, 1993; Affatato et al., 2004; Wolfe et al., 2007). We chose to look at the formation of chromosome aberrations as our endpoint for genetic damage. This approach has been utilized successfully by several groups, including ours, as a marker of disease susceptibility and to elucidate the genotype-phenotype relationships (Abdel-Rahman et al., 2000; Affatato et al., 2004; Wolfe et al., 2007; El-Zein et al., 2006). Materials and Methods Study subjects and collection of blood samples A total of 129 subjects participated in the current study. They were recruited from the staff and student population of the University of Texas Medical Branch (UTMB) in Galveston, Texas. The subjects were volunteers who responded to posted notices throughout the campus, and who were recruited without regard to age, sex, or ethnicity. Individuals were defined as non-smokers if they had smoked less than 100 cigarettes during their lifetime. Individuals were defined as smokers if they had smoked at least 5 cigarettes per day for at least one year prior to enrollment in the study. All study subjects were asked to fill out a questionnaire that provided demographic, occupational, and medical information. Additional information that was collected included details about smoking habits, such as the number of cigarettes smoked per day, preferred brand, duration of smoking, former tobacco use, and use of other tobacco products. All subjects signed a consent that was approved by the UTMB Institutional Review Board and that described the purpose of the study, which is to understand the functional and biological significance of sequence variability in DNA repair genes. Exclusion criteria for all volunteers included a recent acute viral or bacterial infection, a major chronic illness such as cancer or an autoimmune disorder, recent blood transfusion, treatment with mutagenic agents such as chemotherapy or radiotherapy, excessive alcohol consumption, and 20 employment involving exposure to potentially mutagenic chemicals (such as polycyclic aromatic hydrocarbons and nitrosamines) or radiation. Because of these criteria, only apparently healthy volunteers were included in the study to control for potential confounders. After informed consent was obtained, a 50-70 ml blood sample was drawn from each volunteer. A portion of this blood sample was used for DNA extraction for analysis of genetic variations located upstream of the NEIL2 transcriptional start site (identified as the region in chromosome 8 from location 11,663,721 bp to 11,664,692 bp). Another portion was used for RNA isolation and gene expression quantization using absolute quantitative reverse transcription PCR (AQ-RTPCR). The remainder of the blood sample was used for the mutagen-sensitivity assay and, also, for lymphocyte isolation and cryopreservation to provide cells for future studies. RNA extraction and absolute quantitation of copy numbers of NEIL2 transcript by AQ-RTPCR Lymphocytes were isolated from whole blood, as previously described (Affatato et al., 2004; Wolfe et al., 2007; Abdel-Rahman, 2000; El-Zein et al., 2000). Total RNA was isolated from 20x106 frozen lymphocytes using the RNaqueous RNA isolation kit (Ambion, Inc., Austin, TX). RNA samples were initially quantified using a Nanodrop Spectrophotometer (Nanodrop Technologies, Wilmington, DE) and qualified by analysis on an RNA Nano chip. The RNA was further quantified from this chip by using an Agilent 2100 Bioanalyzer (Agilent Technologies; Santa Clara, CA). One µg of total RNA and a NEIL2 mRNA-specific oligo probe, engineered based on data from the NCBI, (Bethesda, MD; NEIL2 accession number NM 145043.1) was used for each reverse transcription reaction. The TaqMan® Reverse Transcription Reagents Kit (ABI, Foster City, CA) was used to generate cDNA for the Q-PCR amplifications. The primers for amplification of NEIL2-specific cDNA were 5’GCTATACACTGCTGGACCAGAGATAC-3’ (forward) and 5’GATGGATCCCAGCTCTGTACAAG-3’ (reverse). The fluorescent TaqMan® probe used for quantification of the NEIL2 transcript was 5’-6FAMCAGGGCTAGGGAACAT-3’. The AQ-RTPCR amplifications (performed in triplicate) 21 were done with 2µl of cDNA in a total volume of 25µl, using either TaqMan MGB probes with the TaqMan Universal PCR Master Mix (ABI) or using SYBR green with the SYBR Green PCR Master Mix (ABI), as specified by the manufacturer. The final concentration of the probe was 250 nM and that of the primers was 900 nM. Absolute quantitation of copy numbers was performed using known amounts of a synthetic transcript of NEIL2 that was generated by amplifying a 1.0 Kb fragment from NEIL2 cDNA in pET 22b using primers 5’- GAATTCATGCCAGAAGGGCCGTTGGTGAG-3’ and 5’- AAGCTTAGGAGAACTGGCACTGCTCTGGC-3’. The PCR conditions were: denaturation 94ºC, 30 sec; annealing 62ºC, 30 sec; 20 cycles; then synthesis 72ºC, 4 min. A TA clone using the TA Cloning Kit (Invitrogen, Inc., Carlsbad, CA) was created with this fragment, and the integrity of the fragment was verified through direct sequencing. The NEIL2 fragment was then sub-cloned into an EcoRI/HinDIII site of the plasmid T7PA. All PCR assays were run in the ABI Prism 7000 Sequence Detection System (ABI) and the conditions were: 50ºC, 2 min; 95ºC, 10 min; 40 cycles: 95ºC, 15 sec; 60ºC, 1 min. Fifteen percent of the samples were randomly selected for a second AQ-RTPCR in order to ensure accuracy of the analysis. For a positive control, we evaluated expression of the XPD gene in the same samples using AQ-RTPCR, since we have previously shown that XPD expression was decreased in smokers relative to non-smokers (Wolfe et al., 2007). In addition, a sub-set of cells from individuals with previously characterized XPD gene expression levels were used as positive controls for the current study. Determination of allelic variants in the 5’-upstream region of the NEIL2 gene A 1113 bp fragment of the NEIL2 5’- upstream region was amplified using PCR primers (upper primer 5’-GGCTCCCCATCCTCCTT-3’ and lower primer 5’GGCCCGCCCTCCCTTCCT-3’). This fragment was sequenced in both directions using the upper and lower primers described above, as well as two internal primers (internal lower primer 5’-TGGCGCCCGGCTAATTTTTGTAT-3’ and internal upper primer 5’TAGCCGGGCGCCAGTAATCC-3’). These sequences were aligned using DNAsis 22 software (Hitachi, Inc., US) and compared with the known sequence from the NCBI database. Genotype and allelic frequencies were determined from these data. Cytogenetic cultures for the mutagen-sensitivity assay Cell cultures were established according to standard procedures (Wolfe et al., 2007; El-Zein et al., 2000; Hill et al., 2005). In brief, aliquots of 1 ml of blood were cultured with 9 ml of RPMI 1640 medium supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin, 10% fetal bovine serum, and 2% L-glutamine (Gibco-Invitrogen, Carlsbad, CA). Stimulation of the peripheral blood lymphocytes (PBLs) was accomplished by the addition of 1% phytohemagglutinin (PHA, reagent grade; Remel, Lenexa, KS). Two cultures were set up in parallel for each study subject; one culture was untreated to represent baseline in vivo chromosome aberration (CA) frequency, and the second culture was used for the mutagen-sensitivity assay. After 46 hours, the cells were centrifuged and the growth medium reserved. The PBLs were then resuspended in 5 ml serum-free RPMI 1640 supplemented with 0.24 mM NNK (98% purity; CAS No 6409191-4, National Cancer Institute, Midwest Carcinogen Repository, Kansas City, MO) and incubated at 37oC in the presence of 5% CO2 for 1 hr. Following NNK treatment, the cells were washed twice with serum-free RPMI 1640, transferred to clean tubes, and resuspended in the original growth medium until harvested. Harvesting was performed at 24 hrs after NNK treatment. Cultures for baseline determination did not receive the NNK treatment. The NNK concentration, exposure duration and harvest time had been established by previous studies (Affatato et al., 2000; Wolfe et al., 2007; Abdel-Rahman et al., 2000; El-Zein et al., 2000; Hill et al., 2005) to produce measurable levels of genetic damage and low levels of toxicity over a period of time that allows the effects of DNA repair to be manifest. 23 Cell culture harvest and cytogenetic analysis for the mutagen-sensitivity assay Prior to harvesting, cells from all cultures were treated with 0.1 µg/ml colcemid (Gibco-Invitrogen) for 1 hr to arrest the cells in metaphase. The cultures were centrifuged and the cells resuspended in hypotonic solution (0.075 M potassium chloride), fixed with Carnoy’s fixative (3 parts methanol/1 part acetic acid, vol/vol), and stored at 40oC. Slides for cytogenetic analysis were prepared, stained with Giemsa, and air dried. Slides were then coded to protect against scorer bias. One hundred metaphase cells on each slide were scored for CA’s, according to standard procedures (International System for Human Cytogenetic Nomenclature), using a Nikon 400 light microscope. Aberrations were recorded as chromosome breaks or chromatid breaks. Chromatid breaks were counted as one break and chromosome breaks as two breaks. The average number of breaks/100 cells was then calculated. Statistical analysis. The Student's t-test, Chi-square, ANOVA, and Aspin-Welch Unequal-Variance Test were used, when appropriate, to compare the mean or median SNP frequencies, between high- and low-NEIL2-expressing groups. Analysis of variance and the Tukey–Kramer multiple comparison tests were used to test differences in CA and NEIL2 expression levels between groups as a function of NEIL2 genotypes. We tested the effects of the SNPs on expression levels using a dominant model. Pearson’s correlation was used to correlate replicates for the AQRTPCR. We also conducted linkage disequilibrium (LD) analysis between the SNPs using Linkage Disequilibrium Analyzer ver. 1.0 (Keyue et al., 2003). The degree of LD among the SNPs was measured using Lewontin’s D′. Statistical tests of the significance (P < 0.05) of LD among these SNPs were conducted using a Likelihood-Ratio test. All statistical analyses were performed using NCSS 2004 (Kaysville, UT). statistically significant. 24 A two-sided P<0.05 value was considered Results Demographics of the study population. Selected demographic information for the study population is presented in Table I. Table I: Selected demographic characteristics of the study population ____________________________________________________________________ N (%) Agea Age range All subjects 129 (100) 41.60 (1.07) 21-73 Sex Male 30 (23) 42.73 (2.21) 23-73 Female 99 (77) 39.95 (1.22) 21-65 Smoking status Smokers 69 (53) 40.10 (1.61) 21-73 Non-Smokers 60 (47) 41.17(1.38) 22-72 Ethnicity White non-Hispanic 92 (71) African American 16 (12) Hispanic 10 (8) Asian 10 (8) Native American 1 (1) ________________________________________________________ a Age in years, expressed as mean (±SE) . The majority of the study subjects were White non-Hispanic (71%). The remaining subjects (29%) were African American, Hispanic, Asian, and one Native American. There were more females (77%) in the study than males. There was no significant difference between the mean ages of the females (mean ± SD=39.95 ± 12.10) compared to the males (mean ± SD =42.73± 12.10). There were 69 non-smokers and 60 smokers. There was no difference between the ages of smokers and non-smokers. Smoking intensity, expressed as pack years (defined as the number of packs of cigarettes smoked per day times the number of years of smoking) ranged between 1 and 75 pack years, with a median of 10 pack years (mean pack years ±SE =16±2.3). There was no significant difference in the smoking habits (number of years of smoking, number of 25 cigarettes smoked per day, and pack years) between the different ethnicities. The age of the participants ranged between 21 and 73 years, with a median age of 42 years. Transcript copy numbers of NEIL2 in isolated human lymphocytes The absolute transcript copy number of NEIL2 in PBL samples from of all the study participants (N=129) was measured using AQ-RTPCR. To verify the reproducibility of our assay, we randomly repeated the quantization of NEIL2 transcript copy numbers for 15% of the samples. A correlation coefficient of 0.88 (P<0.001) between the two series of measurements was obtained. The NEIL2 expression level varied up to 63 fold in the study population, with a range of NEIL2 transcript copy numbers between 429 and 27,183 copies/g total RNA. The mean number of copies ± SE of the NEIL2 transcript in the study population was 3438±397. This variability was not normally distributed. Visual examination of the data (Figure 2) revealed a distribution of NEIL2 expression levels in the population that segregated into two distinct categories. 26 Dot Plot NEIL2 transcript/g total RNA 30000.0 20000.0 High expresser 10000.0 Low expresser 0.0 NEIL2_expressio Variables Figure 2 – Distribution of NEIL2 expression in the study population (n=129). Individuals having higher than 6900 copies/g of total RNA were designated as “high expressers” (12%) and those having less than 4150 copies/g total RNA were designated as “low expressers” (88%). In the first category, which included the majority of the subjects (N=114), the transcript copy numbers were all less than 4150, with a range of 429 to 4139 copies. In the second category, which included 15 subjects (12% of the total population) the NEIL2 transcript copy numbers were all greater than 6900, with a range of 6972 to 27,183 copies/μg of total mRNA (Figure 2). Based on this distribution, all individuals exhibiting an expression value above 6900 copies of NEIL2/μg total RNA were designated as “high expressers” and individuals with a value lower than 4150 copies/μg total RNA were designated as “low expressers”. 27 Association of NEIL2 expression levels with demographic characteristics When levels of NEIL2 expression were evaluated with respect to ethnicity, we observed that average expression levels were significantly higher (mean number of transcript copies ±SE = 3876.34±540.24) in White non-Hispanic subjects (N=92) compared with all other ethnicities combined [N=37; mean±SE=2099.05±214.93, P=0.03; Figure 3 (a)]. Within the White non-Hispanic population, there was also a bimodal pattern of gene expression, where 26% of White non-Hispanics were found to be high expressers [Figure 3 (b)]. This was twice as high as the observed 13% of high expressers found in the group composed of the other ethnicities combined. These results suggest ethnic differences, and imply that copy numbers of NEIL2 expression could be associated with certain ethnic haplotypes. When NEIL2 expression was evaluated with respect to smoking status (smokers vs non-smokers), we observed no significant effect of smoking on NEIL2 transcript copy numbers. The mean number of transcripts ±SE was 3008.98±475.58 in smokers versus 3810.86±615.37 in non-smokers. Similarly, no significant difference in expression was observed with respect to gender (mean±SE for males = 3175 ±707; for females = 3517.564±471.96) or with age (linear regression P value = 0.59). To characterize the frequency of allelic variants in the NEIL2 promoter region, 24 subjects (12 from the high-expresser group and 12 from the low-expresser group) who were matched with respect to age, smoking status, ethnicity, and sex were evaluated. From PBLs of each of these subjects, a 971 bp DNA fragment that corresponds to a location in chromosome 8 from 11,663,721 bp to 11,664,692 bp (a sequence immediately upstream from the NEIL2 coding region) was isolated and sequenced. Notably, this sequence was rich in both previously documented SNPs and predicted cis-elementbinding sites (based on NCBI reports and a query of results obtained from the Transcriptional Element Search System, University of Pennsylvania). 28 Copies NEIL2 transcript/ug total RNA NEIL2 expression by ethnicity P=0.03 # * 5000 4000 3000 2000 1000 0 White non-Hispanic Caucasian N=92 All other ethnicities N=37 Figure 3 (a) Dot Plot NEIL2NEIL2_expression transcript number 30000.0 20000.0 10000.0 0.0 0 1 Non-Hispanic Other ethnicities White_and_other White Figure 3 (b) Figure 3 (a) – Distribution of NEIL2 expression stratified by ethnicity (grey bars). *P<0.05 for comparison between White non-Hispanics and other ethnic groups combined, indicate a significantly higher mean NEIL2 transcript copy numbers in White non-Hispanic subjects. Values are expressed in mean ± SE. Figure 3 (b) Distribution of NEIL2 transcript copy numbers amongst the different ethnic groups. 29 Table II: Single nucleotide polymorphism (SNP) frequency in the study population. Table II : Single nucleotide polymorphism (SNP) frequency in the study population. Report ed range of SNP frequencies d NCBI Reference SNP rs7464968 G>T 11664458 0 to 0.0 5 rs8191515 G>A 11664565 0.0 6 rs8191514 rs8191516 rs8191517 rs8191518 ss74800504 ss74800505 a C>G C>T C>T Position in chromosom e8 11664522 11664641 11664650 C> G 11664670 G>T 11664692 C>A 11664623 Ethnicities represented in the reported frequencies Europea n, Asia n c NR c Minor allele frequency in study population N=24 b Minor allele frequency in high expressers N=12 b Minor allele fre quency in low expressers N=12 b 0.04 0.04 0.04 0.06 0 0.13 0. 14 NR 0.13 0.13 0.13 0.0 2 c 0.04 0.08 0 NR c 0. 00 7 NR 0.02 0.04 0 0. 10 to 0. 30 Europea n,Africa America n n, Asia n c NR 0.33 0.58 0.08 0.10 0.08 0.13 0.33 0.29 0.30 NR c NR c c NR a SNP designation reported in the National Center for Biotechnology Information (NCBI) database under the corresponding SNP accession (minor allele is T for rs7464968, A for rs8191515, G for rs8191514, T for number rs8191516, T for 517, G for rs8191518, T for ss74800504 and A for ss74800505) b . rs8191 N= number of chromosomes evaluated in the study population c NR = not reported . d N Frequencies are expressed as percentages of the minor allele This approach allowed the determination in our study population of the frequency of six SNPs previously reported by NCBI. During the sequencing process of these 24 samples, two new previously unreported SNPs, designated as ss74800504 and ss74800505 (a G>T at position 11664692 and a C>A at position 11664623 of chromosome 8, respectively) were identified, for a total of 8 SNPs evaluated (Table II). We compared the SNP frequencies observed in our study population and the frequencies reported in the NCBI SNP database and found that, while most of the SNPs had comparable frequencies (Table II), the minor allele frequencies for the rs8191516 and rs8191517 SNPs in our study population varied by a magnitude of at least one fold for each of these SNPs compared to that reported in the SNP database. Estimation of linkage disequilibrium (LD) for the 8 studied SNPs in the 24 subjects studied, indicated a 30 significant LD between the newly discovered ss74800505 and rs8191518 (D’=0.73; P<0.05). When categorized as high or low expressers, the minor allele frequencies were not randomly distributed. The rs8191515 SNP was exclusively found in subjects with the low-expresser phenotype, and the rs8191516 and rs8191517 SNPs were only found in high expressers (Table II). SNP rs8191518 was found at a significantly higher (P=0.01) frequency (0.58) in high expressers than in low expressers (0.08). Collectively, these data suggest that SNPs found in the 5’ upstream region of NEIL2, especially the rs8191518 SNP, may differentially modify the regulation of NEIL2 gene transcription. Relationship between NEIL2 expression and mutagen sensitivity In this study population, the baseline (background) in vivo CA frequencies ranged between 0 and 7 breaks per 100 cells (mean + SE = 0.9 + 0.11). Using linear regression analysis, no relationship was observed between the level of NEIL2 gene expression and the baseline in vivo CA. Similarly, no association was observed between baseline CA frequencies and the presence of the SNPs evaluated. Using the mutagen-sensitivity assay we were able to measure sensitivity of an individual to a specific mutagen by treating the lymphocytes with the potent mutagen, NNK. The results indicate that the mean ± SE frequencies of NNK-induced CAs per 100 cells, after controlling for the baseline in vivo level for each subject, was 2.2 ± 0.25, with a range of 0 to 9 breaks/100cells. Using a two-tailed Students t-test, a significant increase in NNK-induced CA was observed in the cells with either the heterozygous or the homozygous variant form of ss74800504 and ss74800505 compared to cells homozygous for the referent allele (Table III). 31 Table III: Relationship between NEIL2 ss74800504 and ss74800505 polymorphisms and mutagen-induced genetic damage ________________________________________________________________ Genotype N mutagen-induced genetic damagea ____ P value____ ss74800504 G/G 14 1.42 (±0.29) G/T or T/T 4 3.00 (±0.91) C/C 9 1.00 (±0.23) C/A or A/A 10 2.60 (±0.45) 0.04* ss74800505 0.01* ___________________________________________________________________________ * Statistically significant (Student’s t-test two-sided P<0.05). P-values are for comparisons between heterozygous and homozygous variant allele carriers and homozygous wild-type. a Data expressed as mean ± SE net increase in mutagen-induced chromosome aberrations per 100 cells. None of the other SNPs studied showed a significant association with mutagen-induced genetic damage. Discussion In the current study, the NEIL2 transcription levels in lymphocytes of healthy volunteers were evaluated. A dramatic variation in the NEIL2 transcription level among individuals was observed. This variability in expression levels (up to 63 fold) is relatively high compared to that observed with other human genes, for example -actin and GAPD(H) and other DNA repair genes, such as XPD and MGMT (Wolfe et al., 2007; Leong et al., 2007; Bustin, 2000; Tanaka et al., 2005). Interindividual variability in the expression levels observed was not correlated with smoking status, as indicated by the lack of association between smoking and levels of NEIL2 gene expression. This finding is in contrast to previous findings from a study with another DNA repair gene, XPD, where 32 XPD transcript levels were decreased in heavy smokers compared to light smokers or non-smokers (Wolfe et al., 2007). In the current study, the transcript levels of both NEIL2 and XPD in a subset of individuals (N=78 of our total population of 129, data not shown) were compared. While a significant decrease in XPD transcript copy numbers in smokers was observed, as expected, no significant change in NEIL2 transcript numbers in relation to smoking status was observed. Several in vitro studies have evaluated the effect of tobacco smoke condensate in various cell types on overall gene expression using micro-arrays. These studies used similar micro-array gene panels and while some of these studies illustrate changes in the expression of other DNA-repair genes, none reported NEIL2 to be responsive to such exposure (Mohrenweiser, 2007; Maunders et al., 2007; van Leeuwen et al., 2005). Maunders et al. (2007) found that exposure of human bronchial epithelial cells to tobacco smoke increased the expression of genes coding for several DNA repair enzymes such as POL and POL, as well as the NER-associated enzyme, GADD 35. Van Leeuwen et al. (2005) showed, in peripheral blood cells, that tobacco smoke exposure did not affect the expression of several genes associated with the repair of DNA damage, such as XRCC1, XRCC5, and ERCC5. But to our knowledge, no studies have illustrated the effects of tobacco smoke exposure on DNA glycosylases, such as NEIL2, in human cells. In the current study, a significant association between levels of expression and ethnicity was observed. Proportionally, white non-Hispanic subjects constituted the majority of the high expressers, and as such, they had significantly higher levels of NEIL2 expression than subjects from all other ethnicities combined. These findings are consistent with reports indicating that gene expression phenotypes vary significantly between different populations (Spielman et al., 2007) and that ethnicity plays a significant role in gene expression patterns as well as in disease risk (Spielman et al., 2007). While the exact mechanisms explaining ethnic variability in gene expression levels are still unclear, we hypothesized that SNPs could, at least in part, contribute to the observed differences in gene expression (Spielman et al., 2007; Lefstin and Yamamoto, 1998). The observed ethnic variability for NEIL2 expression also strongly suggests that 33 high expression of this DNA repair enzyme could be associated with certain haplotypes found predominantly in non-Hispanic white groups. For many genes, the 5’ region upstream of the transcriptional start site is known to contain important motifs that are specific for the binding of transcription-regulating proteins. Inherited differences in these upstream cis-element-sequences (such as the presence of certain SNPs) could, therefore, alter the binding of these transcriptional regulating proteins, resulting in a change in the recruitment or activity of these proteins. This could, in turn, result in an alteration in transcriptional levels, which provides a plausible mechanism that could explain the variations in NEIL2 expression that we observed. In the current study, we evaluated the effect on gene expression of 8 SNPs in the 5’ region of NEIL2 (six had been reported in the NCBI SNP database and two were newly identified in our study). Our data indicate that the rs8191518 SNPs is significantly associated with reduced gene expression levels. The exact mechanism by which this SNP alters expression is still not known. However, we hypothesize that, given that their existence in close proximity to or in areas rich in putative cis-element-sequences as determined by our TESS analysis, they could change the architecture of the DNA binding site, thus altering the affinity of the transcription factor for that site, and subsequently, affecting the transcript level(s) responsive to this factor. Another possibility is that structural changes in the DNA could also create allosteric changes in the transcriptional protein-binding site, which could alter the formation of a complete transcription complex, ultimately leading to altered levels of gene expression, as suggested by Lefstin and Yamamoto (1998). In order to elucidate the effect of the NEIL2 promoter region SNPs on genomic integrity, we characterized the genotype-phenotype relationship using the mutagensensitivity approach. This assay is based on the quantization of mutagen-induced CA in cultured lymphocytes of an individual and it reflects the sensitivity of that individual to the mutagen tested, as well as her/his DNA repair capacity in response to a mutagenic insult (Hsu et al., 1989). Our data indicate that only the two newly identified SNPs had a significant effect on mutagen-sensitivity. 34 The presence in PBLs of the variant ss74800504 or ss74800505 alleles was associated with a significantly higher frequency of mutagen-induced genetic damage when compared to cells with the homozygous wildtype form. This suggests a possible effect of these two newly identified SNPs on DNA repair efficiency. While the exact mechanism(s) underlying this relationship is yet to be determined, it is plausible that SNPs in the NEIL2 promoter could be associated with an alteration in NEIL2 expression levels, or may be in linkage disequilibrium with other functional SNPs in the coding region of the gene, which could result in alterations in NEIL2 protein function. The frequency of the ss74800504 and ss74800505 alleles in our subjects was similar in both high and low expressers, suggesting that these SNPs do not affect expression levels; however they were only evaluated in a relatively small population (N=24). Also, because of the relatively small sample size studied, we could not evaluate the effects of polymorphic combinations on gene expression levels. Additional studies with a larger population are clearly warranted to clarify the observed findings. Such studies would also address the relationship between gene expression levels and NEIL2 protein levels and function. Taken together, our results indicate that variability of NEIL2 expression is, at least in part, influenced by SNPs in the promoter region of the gene, and this variability appears to be affected by ethnicity. Because of the observed mutagen sensitivity associated with both the ss74800504 and ss74800505 SNPs, the potential role of these two new SNPs as risk modifiers for disease susceptibility warrants further investigation. 35 CHAPTER 3: REGULATORY ELEMENTS RESPONSIVE TO OXIDATIVE STRESS IN THE PROMOTER REGION OF NEIL2, A HUMAN DNA GLYCOSYLASE GENE Introduction As described in the previous chapter, NEIL2 expression varies greatly in vivo. Furthermore, at lest one polymorphic site in the promoter region of the gene appears to be related to gene expression. This suggests that NEIL2 expression is governed by regulatory motifs in the 5’ upstream region where single base changes in these motifs could result in modified gene expression. In an effort to characterize the factors in the promoter region that affect the expression of NEIL2, the transcriptional start site was mapped using the random amplification of cDNA ends (RACE) primer extension kit. RACE is a technique commonly used to identify transcriptional start sites (Alberts et al., 2002). It entailed the amplification of the 5’ end of mRNA transcripts using a reverse transcription PCR. Primers were designed to aneal within the mRNA transcript. This allowed for the amplification of the 5’ end of the mRNA. This fragment was then sequenced and resultant sequence was compared to the human genome using alignment software provided by the National Center for Biotechnology. The alignment revealed the exact genomic loction of the first DNA nucleotide that corresponded to the mRNA transcript. We then constructed several luciferase plasmids containing fragments of the NEIL2 promoter region, using the transcriptional start site as a guide to estimate the location of the NEIL2 promoter region. We chose to test these constructs in the normal human lung cell line, MRC-5, for several reasons. First, studies have shown that lung tissue is particularly vulnerable to damage from ROS because of its constant exposure to oxygen as well as air-born environmental pollutants, which can be strong generators of oxidative radicals (reviewed by Langen et al., 2003). Also, the NEIL2 protein has been extracted from MRC-5 cells in other experiments, thus these cells should have the 36 transcriptional machinery needed to drive NEIL2 expression from a transient plasmid (Hazra et al., 2002). These cells have been used for reporting promoter-driven expression using the luciferase reporter assays by other groups (Liao et al., 2006; Wang YP et al., 2006) Finally, MRC-5 cells are an economical and practical choice for studies such as this one where several constructs were tested in numerous conditions. Subsequently, the hypothesis that NEIL2 transcription is responsive to oxidative stress was tested. To induce oxidative stress in our cell line, we chose the persistent H2O2 generator, glucose oxidase (GO). This chemical was used by Das et al. (2005), to induce reactive oxygen species in vitro and to induce the expression of NEIL1expression. Our study identified a positive and a negative regulatory region within the NEIL2 promoter. Expression from the positive regulatory region decreased in the presence of oxidative stress, suggesting that ROS may play a role in NEIL2 expression. Materials and methods Cell culture MRC-5 cells (normal embryonic human lung fibroblasts) were acquired from ATCC (lot no. 3929228; American Type Culture Collection, Manassas, VA) and were cultured in flasks containing Eagle’s Minimal Essential Medium (EMEM, Cellgro Inc., Manassas, VA) with Earle’s basal salts and 2 mM L-glutamine, supplemented with 1.0 mM sodium pyruvate, 0.1 mM nonessential amino acids, 1.5 g/L sodium bicarbonate, 100 U/mL penicillin, 100 g/mL streptomycin, and 10% fetal bovine serum (GibcoInvitrogen, Carlsbad, CA). The cultures were incubated at 37 °C with 5% CO2. Twentyfour hours prior to transfection, cells were harvested from the flasks, counted, and seeded in 6-well plates at a concentration of 3.5x105 cells/well. Mapping of the NEIL2 transcriptional start site The NEIL2 transcriptional start site was mapped by first isolating total RNA from the growing MRC-5 cells. The FirstChoice RLM-RACE primer extension kit (Ambion, Inc., Austin, TX) was used to identify the transcriptional start site according to the instructions of the manufacturer. Basically, the primer extension kit involves ligating the 37 5’ RACE adaptor directly to the 5’ end of the mRNA transcripts. This provides a primer template for sequencing through the adapter and into the mRNA transcript. The sequence found immediately after the adaptor denotes the earliest nucleic acid residue in the transcript, which is the transcriptional start site (Figure 4). Briefly, total RNA was isolated from MRC-5, cells and 10 g of the total RNA was treated with calf intestine alkaline phosphatase (CIP) and then treated with tobacco acid pyrophosphatase (TAP) to specifically modify the full length mRNA for adapter ligation. The 5’ RACE adapter (included with the primer extension kit) was ligated to the 5’ end of the full length mRNA, which provided a template for primer extension during the subsequent reverse transcription and PCR amplifications. In the first reaction, the 5’ RACE outer primer (also included with the kit) was used in combination with the NEIL2 gene-specific primers (RACE1, 5’-TCTTCCGAGTGCCCGAGGTG-3’ and RACE2, 5’ - GGGGTTGTCTCCGCCGTTC - 3’) that were designed based on GenBank mRNA (NM_145043) sequence data. Then a second PCR reaction was used to amplify the region using the same genespecific primers and the 5’ RACE inner primer (included with the extension kit). Products of this latter reaction were sequence-verified using the ABI Prism 3100 Sequence Detection system (Applied Biosystems, Foster City, CA) and BLASTn software (NCBI, Bethesda, MD). Following identification of the transcriptional start site through sequencing through the adapter region to the junction of the NEIL2 transcript, the transcription element search system (TESS, University of Pennsylvania), which allows the identification of functional cis-acting binding sites within the DNA strand (Schug and Overton, 1997), was subsequently used to search for putative cis-elements located near the transcriptional start site. 38 Figure 4: RACE methodology mRNA cap AAA… mRNA transcript CIP AAA… TAP 5’OH AAA… adapter Ligation AAA… Gene-specific sequencing primer AAA… Adapter-specific sequencing primer Cloning of the NEIL2 5’- upstream region into luciferase reporter vectors The results obtained from the TESS analysis guided the design of NEIL2 promoter constructs within a 1871 bp region encompassing from bp -1161 to +710 of the NEIL2 gene (Figures 5 and 6). 39 -1161 Untranslated region Coding region +710 +1 -1161 Chomosome 8 – 11664692 Chomosome 8 – 116682263 pSTEP1 -1161 +710 pSTEP2 -513 +49 p1200 -1161 -1161 p1200A -702 +710 pSTEP3 +710 +90 -661 p1200B -104 -206 p1200C +90 Figure 5: Schematic representation of the NEIL2 gene and the relative locations of the promoter fragments isolated, cloned, and sub-cloned into luciferase reporter vectors. The identified transcriptional start site is designated as +1. The first and longest construct encompassing this region (1871 bp) was designated as pSTEP1 and was found to include several putative transcriptional binding sites (Figure 6). The term STEP was used because we used a step-wise truncation pattern to reduce the size of the DNA fragment contained in each of our clones. The second construct, the pSTEP2 construct (1223 bp, from bp -513 to +710) was then designed as a 40 (-1161) AP-1 GACGGTGGGCTGGGCTTGTTTCCCTGTGCTGACCAAGTCCTTCCCCAGGGACCCCTCAGCTAG NFkB AP-1/CREB GGTCCATGGTGACTTCACCCCAGCCTATTTCCTGAACACCTGCTGGGGAGTTCCCCAGACTTTC CTCCCAACTTCCTGGAAAGTGCTGGCTCCCCATCCTCCTTCGAGAACTAAAGACAAGAAAACA TATGTATCTTTAAACAGCCTATTGTTTTATCTCTTTTATTCTATGAAATAAAAAATTTAATACA CAAATACTTTAAGCAAAATTTGCTTATAGTAAAAATTCAAGTAATAAAATTCAAATAATAGAA AGCAAAAAAATCCCACCCGTTCAACATTTCACGAGTCTTGAACACCTGTTTCCTTTGCACCCG ACTTCATTAGGCTCGGAGGCCAAGCAGAACCCACAGAAAGGCTTGCCATTGTGGGAACACCC AGGCTGTCCCATTCACACACCCATTGACTTTTCCCTATAACTGTCTCCTTGAGACAAGAAATGT TCCAAAAGGGCCGGGTGCGGTGGCTCACGCCTGTAATCCCACCACTTTGGGAGGCCGAGGAG GGCGGCTCACCTGAGATCAGGAGTTCAAGACCAGCCTGGCCAACATGGTGAAACCCGGTCTC TACTAAAAATACAAAAATTAGCCGGGCGCCAGTAATCCCAGCTACTCGGGAGGCTGAGGCAG GAGAATGGCTTAAGCCCAGGAGGCGGAGGTTGCAGTGAGCCGAGATCGCGCCACTGCACTCC AP-1 AGCCTGGGTGACACAGCGAGACTATCTCCCCCCGCAAAAAAAAAAAGTTTCCAAGAGCGTCT CTCCAATGTTAGTCCCAGCCTGCACCCTTCTTCCATCCCTGCTTTCACTCGCGACCCTCTAATG CAGCCCTGCCTCCAGCGACCCCCGGGATGGAACCCCTCGCCCACCCACAAAAGAGCAGCCTC GACCTAGACCCACTTTCCAGGGAATGAGCCCTGCGCCTGCGCGCACCCCGCCACCCCCTCCAG NFkB SP1 GCCGGAGTCCACGCCCACTTGGGGGAGGAGCCCCGCAGCCTCCACCTACAGGGGCGTCCCCT AP-1/NFkB AAGGGGACGGAGGCCGCATGGGCCGCCGAGCCGGGAAATCTCCGCCCCCAGCTGGAGCGGCT USF SP1 +1 GTGCGGGCTGCGTAGCGGTGCTGGGTCGGGCCGACGTGCCACCCACCCGGAGCCGGTGAGTG CAGCCGCCCGCCCTCCGGTAGATCTGCGGCCTGGCGGAGAAGTCGGGAGGGGACAGGAAGGG AGGGCGGGCCCCGGGCCCTCCTCCGTCTCAGCCGCCTGCGGAGGTGCTGCCCACGCCTGGAGG CCCCCACTGACCCTCAGACCCGCGTCTGCGCCCCTCTCCCCGCACCCCGAGGCAGAGTTGGGA AAGCAGTGGTCTTAGACCCCCCACCTCGGGCACTCGGAAGAGAACGGCGGAGACAACCCCTC CTCTTCCCTGGCTGGCGCAGCGCCAGCCTCGAGCTCCTCGGTAGCCCCCGGGCAGGGAGGGCC GGAGGGTGGGCGCGGCATCTTCAGCGACTCTTCGAAGTCCCTTCCGCGTCTCATCTTTCAAGG CTGTTGCAGAGGCGGCTTGCTTCCCACCTGTCCATCTCCATAAAAATCCCTAAACGAAACATG AP-1/CREB CCCACGTGTCCGGAGATTTTCAGGACTTGGTGCATTTCAGATGAAGGCTTTTCCAGAAGCTTCC CCGTAGAAGAGGATCAGGCATCCAACTGGTTAAGGTCAGCAGCGTTTGGCACGTCTCCTTCCA GCCTGGCGGTTTTGTCAGGATTCCCTGGGGAGTGTCTGGAAAGCCTGATGAGGGGAAATAGTA CATCTCAGCGAATCGGCACCAGCGAGTGTAAGATGCGCGTTATTGAATGTG(+710). 41 Figure 6: The DNA sequence, encompassing the pSTEP1 fragment, that includes the NEIL2 gene sequence flanking 5’ upstream and 3’ downstream of the identified transcriptional start site (+1). Putative consensus sequences for binding to regulatory factors are activating protein – 1 (AP-1; highlighted), cAMP response element-binding (CREB; italicized), nuclear factor –kappa B (NF-B; underlined), and polyomavirus enhancer A factor 3 (PEA3; bold and italicized). truncation of pSTEP1 (Figure 5), which eliminated several putative regulatory elements, including an NFB site (see Figure 6). The third construct we designed, the pSTEP3, contained a region 3’ of pSTEP1 (661 bp, from +49 to +710). This pSTEP3 construct was designed to include the downstream putative transcriptional cis-elements of pSTEP1. As shown in Figure 4, we also designed the construct p1200 containing only the 5’ portion of pSTEP1 (1250 bp, from bp -1161 to +90). This p1200 construct was designed so that it retained the putative transcriptional elements upstream of the transcriptional start site. As such, each of these constructs was engineered to contain a unique set of cis-elements (Figure 6). This design allowed us to evaluate the independent contribution of each region in driving NEIL2 expression. The 5’ gene region of NEIL2 was amplified from genomic DNA through PCR. We used primers that were specifically designed to amplify the regions of interest (Table IV). Table IV - Primer sequences for luciferase constructs Luciferase construct Forward primer Reverse primer pSTEP 1 5' - GAGCTGGTGGGCTGGGCTTGTTT - 3' (5' - CACATTCAATAACGCGCATC - 3' pSTEP 2 5' - TAGCCGGGCGCCAGTAATCC - 3' 5' - CACATTCAATAACGCGCATC - 3' pP1200 5' - GAGCTGGTGGGCTGGGCTTGTTT - 3' 5' - GAAGCTTTCCCCTCCCGACTTCTC - 3' p1200A 5' - GAGCTGGTGGGCTGGGCTTGTTT - 3' 5' - CTTGTCTCAAGGACACAGTT - 3' p1200B 5' - CACCCATTGACTTTTCCC - 3' 5' - TGTAGGTGGAGGCTGCGG - 3' p1200C 5' - TTTCCAGGGAATGAGCCCT - 3' 5' - GAAGCTTTCCCCTCCCGACTTCTC - 3' 42 The resulting fragments were then cloned into the pCR2.1 vector, using the TA Cloning Kit (Invitrogen, Inc., Carlsbad, CA), and then sub-cloned into the KpnI/EcoRI restriction sites in the promoter-less luciferase expression plasmid, pGL3-Basic (Promega, Inc., Madison, WI), to create the three plasmids: pSTEP1, pSTEP2, and p1200. Construct pSTEP3 was created by cloning a 661 bp fragment of the region from +49 to +710 into the BglII site of the pGL3-Basic vector. Sub-clone constructs of the p1200 and pSTEP3 fragments In addition to the constructs described above, three fragments were amplified from the p1200 construct (see Figure 5) using the primers shown in Table IV. The fragment encompassing the 5’ region of the p1200 insert (from -1161 to -661) was used to create the p1200A construct. The central fragment (from -702 to -104) was used to create the p1200B construct. The 3’ region (from -206 to +90) was used to create the p1200C construct. In addition to these p1200 sub-clones, to evaluate the characteristics of the pSTEP3 fragment (661 bp, from +49 to +710), we cloned this fragment into a pGLcontrol vector containing the strong, ubiquitous SV-40 promoter. The pSV40-STEP3 construct was created by ligating the BglII fragment from pSTEP3 into the BglII site of the pSV40 vector. The integrity of the cloned fragments was verified by direct sequencing. Plasmid DNA used for transfections was isolated using the Qiagen EndoFree Maxi-prep (Qiagen, Inc., USA). The stock DNA extract was diluted in endotoxinfree water to a final concentration of 1g/uL prior to transfection. In order to ensure plasmid integrity, all plasmid preps were performed three days prior to transfection. Transient transfection of MRC-5 cells with NEIL2 promoter constructs and luciferase assay MRC-5 cells were transfected with the constructs described above for 4.5 hours in a mixture of 1.0µg luciferase and 0.5 µg Renilla DNA/well at a Lipofectamine (Invitrogen, Inc.) concentration of 6 of Lipofectamine/µg plasmid DNA. Transfection efficiency was tested using the Dual Luciferase Reporter System (Promega, 43 Inc.). After transfection, cells were allowed to recover for 48 hours and then were harvested in 1x Passive Lysis Buffer (Promega, Inc.). Luciferase reporter gene expression was detected using the Luciferase Detection Kit (Promega, Inc.). Briefly, cells were washed in phosphate buffered saline and incubated in 200 μl of cell lysis buffer. Luciferase activity was measured according to the manufacturer’s instructions and detected on a GENios Pro micro-plate reader (Tecan, Inc., Durham, NC). The total cellular protein concentration was determined using a Bradford-based assay from Bio Rad (Bio Rad Protein Assay, Bio-Rad Laboratories, Hercules, CA). Total recoverable cellular protein concentration was determined by calibration relative to a standard curve generated using known concentrations (between 0-24 mg/ml) of aqueous bovine serum albumin. Luminescence was measured in relative light units (RLUs) per L. The relative luciferase activity in each sample was normalized to the total concentration of protein. Each experiment was repeated at least five times. Response of the p1200C and NEIL1 construct to oxidative stress MRC-5 cells were transfected with either the p1200C construct (the fragment from bp -206 to +90) or the pNEIL1 luciferase construct and treated with 100 ng/ml of the cellular oxidant glucose oxidase (GO; Roche, Inc. Basel, Switzerland). Glucose oxidase is a strong generator of cellular hydrogen peroxide (H2O2) which produces an environment conducive to oxidative DNA damage (Das et al., 2005). Prior to treatment, cultures were allowed to recover in fresh medium for 24 hrs after transfection. Cells were continually treated with GO, and luciferase expression levels were determined at 1, 6, and 12 hrs after treatment. The experimental conditions used did not affect cell viability, as determined by trypan blue exclusion. Statistical analysis The Student's t-test and ANOVA were used, when appropriate, to compare the means between treatment groups. A two-sided P<0.05 was considered statistically significant. A Tukey’s test was used when appropriate to designate different groups. All statistical analyses were performed using NCSS 2004 (Kaysville, UT). 44 Results Mapping of the NEIL2 transcriptional start site and identification of ciselements in the NEIL2 promoter. Using the FirstChoice RLM-RACE kit, we determined that the guanine residue at location 11664692G on chromosome 8 was the transcriptional start site (designated as +1 in Figures 5 and 6). The TESS software (Schug and Overton, 1997) was used to identify, in this region, several potential cis-elements for transcription factors, including a nuclear factor-kappa B (NF-kappaB) protein, a cAMP response element-binding protein (CREB), and an AP-1 site (a heterodimer formed from C-jun and C-Fos) (Figure 6). Partial characterization of the NEIL2 promoter region. As shown in Figure 7A, the largest construct of the NEIL2 promoter fragment pSTEP1 (the fragment from bp -1161 to +710) drove minimal gene expression. we then used the TESS software to search the sequence for sequence areas that were enriched in transcriptional elements. The results of this search showed that the region from bp -1161 to +90 had several transcriptional binding motifs. Therefore, the p1200 construct (from bp -1161 to +90), which contained the first 1251 bp of the -1161 to +710 fragment, including the transcription start site was designed. This was equivalent to deleting the pSTEP3 fragment (bp +49 to +710) located in the -1161 to +710 fragment. The -1161 to +90 fragment (p1200) showed a 5-fold higher expression level compared to the -1161 to +710 fragment (pSTEP1), suggesting that it contained positive regulatory elements (Figure 7B). A plausible reason for the higher expression from the -1161 to +90 compared to the longer -1161 to +710 fragment is that the missing +49 to +710 fragment contains negative regulatory elements that repress the induction of NEIL2 expression observed with the pSTEP1 construct. To further evaluate potential repressive characteristics of the +49 to +710 fragment (pSTEP3), it was cloned into a pGL-control vector containing the SV-40 promoter (Figure 8). 45 A -1161 +710 -513 LUC pSTEP1 LUC pSTEP2 LUC pSTEP3 +710 +49 +710 -1161 +90 LUC p1200 B pSTEP1 pSTEP2 pSTEP3 * P1200 0 50 100 150 200 250 300 Relative light units/g total protein Figure 7: A, Luciferase constructs containing DNA fragments of the 5'regulatory region that were cloned upstream of the luciferase-coding sequence; B, Promoter activity (determined as described in Materials and Methods and show in means ± SE). * P < 0.05 compared to pSTEP1. 46 A +49 pSTEP3 fragment SV-40 LUC SV-40 LUC +710 pSV40 pSV40-STEP3 B pSV40 pSV40-Step3 * 0 1000 2000 3000 4000 5000 6000 7000 RLU's/mg total protein Relative light units/g total protein Figure 8: A, Luciferase constructs containing the pSTEP3 DNA fragment inserted 5’ of a SV-40 promoter; B, Promoter activity relative to a pSV-40 control vector is shown in relative light units (RLUs) per mL, normalized to total protein in the sample. *P<0.05. 47 Inserting the +49 to +710 fragment proximal to the SV-40 promoter (pSV40-Step3) resulted in a 91% decrease in SV-40-driven luciferase expression (Figure 8B), further confirming the presence of repressive elements in this region. Further characterization of the key regions responsible for positive regulation of the NEIL2 gene was completed by sub-cloning segments of the -1161 to +90 fragment (p1200) into three constructs (p1200A, p1200B, and p1200C; Figure 9A). A -1161 +90 -1161 LUC p1200 LUC p1200A LUC p1200B LUC p1200C -661 -702 -104 -206 +90 B P1200 ** p1200A * p1200B p1200C 0 50 100 150 200 Relative light units/g total protein 48 250 300 Figure 9: A, Luciferase sub-clone constructs containing fragments (A, B, and C) of the p1200 DNA fragment inserted upstream of luc-coding sequence; B, Promoter activity (determined as described under Materials and Methods in relative light units/g total protein). *P < 0.05 compared to p1200C. **P<0.05 compared to p1200B. While the relative promoter activity of the p1200C construct (containing the fragment from bp-206 to +90 and the transcriptional start site) was comparable to the relative promotor activity of the -1161 to +90 fragment (p1200), the relative promoter activity of the p1200A construct (the fragment from bp -1161 to -661) was 5-fold lower than that of p1200C (P<0.01) and significantly lower that p1200B (P<0.05). In addition, the promoter activity of the fragment from bp -702 to -104 (p1200B) was 3-fold lower than that of p1200C (P<0.01; Figure 9). These observations are consistent with the p1200C fragment containing the positive regulatory region of the NEIL2 promoter. Response of the p1200C construct (-206 to +90 fragment) and pNEIL1-luc to oxidative stress Because the fragment from bp -206 to +90 (p1200C) drove the highest level of luciferase expression and, presumably, had positive regulatory elements for NEIL2, the next step was to examine whether this fragment was responsive to oxidative stress. The 206 to +90 fragment responded to GO treatment, with a rapid and significant decrease of 29% in luciferase expression from 1 hr (P=0.04) to 6hr (P<0.01) of continual treatment (Figure 10 followed by a recovery to untreated levels at 12hr. 49 4 3.5 3 * * 2.5 2 1.5 1 * * 6hr 12hr 0.5 0 0hr 1hr 6hr p1200C 12hr 0hr 1hr pNEIL1 Figure 10: Luciferase reporter activity in cells transfected with the p1200C and pNEIL1 constructs and then treated with glucose oxidase (100 ng/mL) for 1, 6, and 12 hrs. Comparisons are between GO treated at 1, 6, and 12 hrs vs. untreated. *P < 0.05. At 12 hr, expression completely recovered to the same level as the untreated (Figure 10). After no response at 1hr, the pNEIL1-luc construct had a progressive, significant decrease in expression, from 6hr to 12hr. Discussion A growing body of evidence indicates that NEIL2 plays an important role in transcription-coupled DNA repair and, potentially, in cancer susceptibility (Hazra et al., 2002; Hazra et al., 2003; Broderick et al., 2006). In view of the relative importance of NEIL2 in maintaining genomic stability, this study initially characterized the NEIL2 promoter and mapped the transcriptional start site of this gene. To our knowledge, the promoter region of the human NEIL2 gene has not been characterized before, nor have any attempts been made to identify the transcriptional start site of this gene. The Luciferase Reporter System was used to evaluate the expression characteristics of several fragments of the NEIL2 promoter region. Our evaluation of the expression activities associated with these fragments revealed that the 1871 bp fragment from -1161 to +710 50 (pSTEP1 construct) did not drive expression in MRC-5 cells, even though this fragment contained the transcriptional start site as well as several putative transcriptional element binding sites. These findings suggested that this fragment could contain negative regulatory regions that suppress expression. In fact, sub-cloning of this fragment demonstrated that it contains both positive and negative regulatory regions that seem to be affecting gene expression. A positive regulatory region was localized in the fragment encompassing bp -206 to +90 that includes the transcriptional start site (the p1200C construct). However, the expression from this region was found contingent upon its being isolated from the adjacent fragment (+49 to +710, pSTEP3), suggesting the presence of silencing elements in this region. In order to further characterize the gene-silencing potential of this region, the +49 to +710 fragment was inserted next to the SV40 promoter of the pGL3-control vector to test whether it would repress the expression of this promoter. The SV40 promoter is commonly used in the pGL3-control vector as a positive transfection control because it promotes strong reporter-gene expression in various mammalian cell types (Wildeman, 1988). As such, repressing its expression would, presumably, require robust repression elements. We observed a 91% reduction in SV40-driven luciferase expression associated with this fragment, consistent with it, containing repression elements that negatively regulate NEIL2 gene expression. This study further showed that the fragment from bp -1161 to +90 (p1200) drove substantial luciferase expression. As such, this fragment likely contains positive regulatory elements. Most of the p1200 fragment was 5’ of the transcriptional start site. The majority of the well-characterized gene regulatory elements are usually found within a region that is approximately 2000 bp upstream of a gene transcriptional start site (Mottagui-Tabar et al., 2005). Indeed, TESS analysis indicated the presence of multiple putative cis-binding sites for various transcription factors within the -1161 to +90 fragment, particularly within the -206 to +90 region, including NFB and AP-1 sites that are known to be responsive to ROS (Muller and Gawlik, 1997). The results from the experiments comparing the three constructs (p1200A, p1200B, and p1200C), each 51 containing different regions of the -1161 to +90 insert (Figure 5, clearly indicate that the 206 to +90 fragment contained essential positive regulatory elements. Because the NEIL2 protein repairs oxidative DNA damage, we hypothesized that NEIL2 expression would be responsive to ROS. Consistent with this hypothesis, these results indicate that GO treatment of MRC-5 cells transiently transfected with the p1200C plasmid (the -206 to +90 fragment) was associated with a down-regulation of expression at the 1hr and 6hr time points (Figure 10 These results are consistent with observations of other investigators who identified other genes whose transcription was down-regulated by ROS, including genes of other BER proteins such as OGG1, cytokines, and TNFα in T-cells, as well as pro-inflammatory genes in human chondrocytes (Sun and Oberley, 1996; Morel and Barouki, 1999; Mathy-Hartert et al., 2003). These investigators have suggested that such transcriptional down-regulation appears to be due to the inhibition of particular transcription factors by oxidative stress (Sun and Oberley, 1996; Morel and Barouki, 1999; Mathy-Hartert et al., 2003). Specifically, trans-activation by the Sp1 factor, the E26 transformation-specific (ETS) transcription factor, and the upstream stimulatory factor (USF) are known to be inhibited in the presence of ROS (Morel and Barouki, 1999). For several of these factors, protein/DNA binding is inhibited by the abnormal protein structures that are induced by an oxidative environment (Muller and Gawlik, 1997). It is interesting to note that there are putative USF and several putative SP-1 sites in the -206 to +90 fragment (P1200C), located upstream but close to the transcriptional start site (Figure 6. These sites are not identified either in the -1161 to 661 fragment (p1200A) or in the -702 to -1104 (p1200B) fragment. A plausible explanation for our findings with the p1200C construct could be that the oxidative conditions induced by GO resulted in the inhibition of the binding of transcriptional binding proteins to the DNA, thus reducing luciferase expression in cells transfected with the p1200C construct. Additional studies are clearly needed to further explain the observed response following GO treatment. It should be noted, however, that a recent study by Das et al. (2007) indicated that NEIL2 protein activity is enhanced in the presence of ROS (Das et al., 2006). This suggests that NEIL2-related repair of oxidative 52 DNA damage could be mediated through induction of protein activity, rather than by the induction of transcription. Expression studies involving the NEIL1 promoter by Das et al. (2006) indicate that NEIL1 expression was induced by a short (1 hr) exposure to 100 ng/mL GO. In the current study, the pNEIL1-luc construct was used as a positive control for oxidative stress. Interestingly, in our study the NEIL1 promoter-driven expression decreased with prolonged exposure to GO (Figure 10 A possible explanation for the observed differences in results could lie in the observation that the Das et al. study (2006) used acute exposure to GO whereas we used the prolonged exposure to GO. It has been well established that ROS act as intracellular messengers, inducing enzymatic cascades that affect a variety of biochemical processes; however, the length of exposure determines the pathways that are involved (Verweih and Gringhuis, 2002). In the review by Verweih and Gringhuis (2002), they list many different proteins that are activated during acute and chronic exposure to ROS in lymphocytes. These are proteins involved in signaling various enzymatic cascades, including those involved in transcriptional regulation. During acute GO exposure, NEIL1 expression is influenced by binding of the AP-1 transcriptional activator to the NEIL1 promoter region; however, the binding factors that could affect NEIL1 transcription during chronic exposure are unknown (Das et al., 2006). One possible explanation for the gradual decrease in NEIL1 transcription seen in our study is that the chronic GO exposure that we used induces repression elements of the gene. Also, NEIL1 expression is known to be cell cycle dependent, with highest expression during S phase (Hazra et al., 2002). It is widely known that H2O2 is a potent intercellular messenger. It has also been shown to induce cellular proliferation in vitro by orchestrating S phase entry (Manasija-Radisavljevic and González-Flecha, 2003). Thus, a plausible explanation for the decrease in pNEIL1 luciferase driven expression is that the persistent H2O2 created by the GO is signaling the cells to move from S-phase and into G2/M phase. In this way, pNEIL1 expression would decline with the change in cell cycle. These explanations seem plausible and open new avenues of future studies to examine the mechanism NEIL1 and NEIL2 expression. 53 We also note that NEIL2-driven luciferase expression from the p1200C construct, when exposed to GO, resulted in a decrease and then a recovery of expression over time (Figure 10 We suggest that the p1200C fragment contains DNA binding motifs for transcriptional regulating proteins that are temporally responsive. Mauders et al. (2007) showed that tobacco smoke, which is an ROS generator, created a temporal variation in the expression of genes involved in the activation of trans-acting proteins. Expression of both the SMAD4 and FOS proteins exhibited little or no change in expression at 1 hr after exposure, then, there was a boost in expression at 6hr. At 24 hr after exposure, expression of these proteins decreased (Mauders et al., 2007). Future studies will help to elucidate the trans-acting proteins involved in NEIL2 expression. This initial down-regulation and then recovery by this NEIL2 promoter region could indicate cellular adaptation of persistent oxidative stress. If this is so and we consider that NEIL2 expression is not cell cycle regulated, we can suggest that the role of NEIL2 protein in oxidative DNA repair is that it is responsible for maintaining genomic stability under normal circumstances. Furthermore, this suggests that the induction of NEIL1 transcription observed by Das et al. (2006) is in response to acute and excessive oxidative stress. These observations denote yet another plausible difference in the roles of NEIL1 and NEIL2 proteins in DNA repair. In conclusion, this study is the first to provide an initial characterization of the NEIL2 promoter and identifies important genetic factors regulating NEIL2 expression. Potential regions in the promoter of the gene that could affect NEIL2 transcription levels and that are responsive to oxidative stress were identified. The results provide novel insight into the inherent genetic mechanisms that could influence interindividual differences in expression and, accordingly, in DNA repair capacity. This study also identifies gaps in knowledge that need to be addressed by future studies. A particularly vital issue is the biological effect of down-regulation of NEIL2 in response to oxidative stress, given the potential importance of this protein in transcription-coupled DNA repair. In rapidly proliferating tissues, reduced NEIL2 transcription could lead to increased DNA damaged from oxidative stress and genomic instability. 54 55 CHAPTER 4: NEIL2 GENE EXPRESSION IS INFLUENCED BY POLYMORPHIC SITES AS WELL AS AN NF-KAPPAB BINDING MOTIF IN THE PROMOTER REGION Introduction Previous chapters have described inherent and genetic factors which regulate NEIL2 gene transcription. The NEIL2 promoter was discovered to have a unique structure containing a positive regulatory region from -206 to +90, as well as a negative regulatory region from +49 to +710 (Figure 8and 9. Also, SNPs within the positive regulatory region of the NEIL2 promoter were determined to be associated with increased sensitivity to mutagenic environmental agents, and were found to be associated with NEIL2 gene expression levels in humans (Table II and III). In addition, we hypothesized that SNPs proximal to key regulatory regions impact gene regulation and DNA repair, especially in the presence of cellular stress. To test this hypothesis, we engineered single base pair modifications into luciferase constructs containing fragments of the NEIL2 promoter region using site directed mutagenesis. These constructs were transfected into MRC-5 cells and luciferase expression was measured. Materials and Methods In silico search for putative transcriptional binding sites The Transcriptional Element Search System (TESS) software (Schug and Overton, 1997) was used to predict how single base changes corresponding to the newly discovered ss74800504, ss74800505 SNPs, and the rs8191518 SNP would eliminate existing, or create additional, cis-regulatory elements in the NEIL2 promoter sequence. The weighted matrix function of TESS was used to identify essential base changes that were predicted to abolish existing cis-regulatory elements. This information was used to target specific base pairs for site directed mutagenesis. This technique allowed testing 56 the effect of base changes on NEIL2-promoter-driven expression under two circumstances: (1) in the presence each SNP(s), and (2) in the event of a mutation that was predicted to eliminate the binding of strong trans-acting factors. Site directed mutagenesis and luciferase expression constructs pSTEP1 mutations Plasmid pSTEP1 was constructed, as previously described in Figure 4 and 6. Briefly, pSTEP1 was created by cloning a 1871 bp fragment of the NEIL2 promoter region (-1161 to +710) into the multiple cloning site of plasmid pGL3-basic luciferase reporter vector (Promega, Inc., Madison, WI). The plasmid pSTEP1 contained a fragment of the NEIL2 promoter from base pair -1161 to +710, including previously described positive and negative regulatory regions (Figures 7-9 For the pSTEP1 construct, the Quickchange Site Directed Mutagenesis Kit was used as per manufacturers’ instructions (Stratagene, Inc., La Jolla, CA), to introduce three, independent mutations. These mutations corresponded to the SNPs rs8191518, ss74800505, and ss74800504 (Table V). Table V: Forward oligo primers used to introduce point mutations into luciferase plasmids. Primer name rs8191518 NFkB-1 (-104 G to C) ss74800505 ss74800504 Briefly, mutagenesis Sequence – Single base change indicated in bold TCTGCGGGCTGCGTAGGGGTGCTGGGTCGGGCCGAC GCCTCCACCTACAGGCGCGTCCCCTAAGGGG CATGGGCCGCCGAGACGGGAAATCTCCGC TGGGTCGGGCCGACGTTCCACCCACCCGGAGC was completed by denaturing the plasmids and then re-annealing them in the presence of oligo primers (Table V), each containing one single base mutation (underlined and in bold) at the intended site. These oligos acted as primers for subsequent PCR cycles, creating a replicon that contained the intended site of mutation. The resultant double stranded plasmid was then digested with restriction endonuclease DpnI to selectively digest the bacterial plasmid template from the synthetic replicon. 57 This plasmid was then transformed into competent E.coli cells for amplification of the double-stranded plasmid, containing the altered base pair. Each point mutation was verified by direct sequencing. In this way, three sub-clones of pSTEP1 were created, each containing single base changes corresponding to SNPs ss74800504, ss74800505, and rs8191518 (Table VI). Table VI – point mutations in pSTEP1 or p1200C constructs. Constructed Plasmids pSTEP1 with one base change rs8191518, ss74800505, ss74800504 pSTEP1 with two base changes ss74800505 + rs8191518, ss74800505 + ss74800504, ss74800505 + (-104 GtoC) p1200C with one base change ss74800505, ss74800504, (-104 GtoC) p1200C with two base changes ss74800505 + rs8191518, ss74800505 + ss74800504 ss74800505 + (-104 GtoC) p1200C mutations Plasmid p1200C was constructed by using PCR primers to isolate a 296 bp fragment from the pSTEP1 (-1161 to +710) insert and cloning it into the multiple cloning site of the pGL3-basic luciferase reporter vector (Figure 9. The p1200C plasmid contained the putative, minimal, positive regulatory region encompassing base pair -206 to +90 (Figures 9. The Quickchange Site Directed Mutagenesis Kit was used as per manufacturers’ instructions (Stratagene, Inc., La Jolla, CA), to introduce three independent mutations into p1200C. Two of these mutations corresponded to the SNPs ss74800504 and ss74800505 (Table V). In addition to these changes, a single base change at a location 104 bp 5’ of the transcriptional start site (denoted as -104 GtoC), was 58 predicted to abolish the binding sites for the NF-kappaB and Sp-1 transcription factors. Therefore, this base change was also engineered into the p1200C plasmid (Table VI; Figure 11 59 (-1161) AP-1 GACGGTGGGCTGGGCTTGTTTCCCTGTGCTGACCAAGTCCTTCCCCAGGGACCCCTCAGCTAG AP-1/CREB NFkB GGTCCATGGTGACTTCACCCCAGCCTATTTCCTGAACACCTGCTGGGGAGTTCCCCAGACTTTC CTCCCAACTTCCTGGAAAGTGCTGGCTCCCCATCCTCCTTCGAGAACTAAAGACAAGAAAACA TATGTATCTTTAAACAGCCTATTGTTTTATCTCTTTTATTCTATGAAATAAAAAATTTAATACA CAAATACTTTAAGCAAAATTTGCTTATAGTAAAAATTCAAGTAATAAAATTCAAATAATAGAA AGCAAAAAAATCCCACCCGTTCAACATTTCACGAGTCTTGAACACCTGTTTCCTTTGCACCCG ACTTCATTAGGCTCGGAGGCCAAGCAGAACCCACAGAAAGGCTTGCCATTGTGGGAACACCC AGGCTGTCCCATTCACACACCCATTGACTTTTCCCTATAACTGTCTCCTTGAGACAAGAAATGT TCCAAAAGGGCCGGGTGCGGTGGCTCACGCCTGTAATCCCACCACTTTGGGAGGCCGAGGAG GGCGGCTCACCTGAGATCAGGAGTTCAAGACCAGCCTGGCCAACATGGTGAAACCCGGTCTC TACTAAAAATACAAAAATTAGCCGGGCGCCAGTAATCCCAGCTACTCGGGAGGCTGAGGCAG GAGAATGGCTTAAGCCCAGGAGGCGGAGGTTGCAGTGAGCCGAGATCGCGCCACTGCACTCC AP-1 AGCCTGGGTGACACAGCGAGACTATCTCCCCCCGCAAAAAAAAAAAGTTTCCAAGAGCGTCT CTCCAATGTTAGTCCCAGCCTGCACCCTTCTTCCATCCCTGCTTTCACTCGCGACCCTCTAATG CAGCCCTGCCTCCAGCGACCCCCGGGATGGAACCCCTCGCCCACCCACAAAAGAGCAGCCTC -206 GACCTAGACCCACTTTCCAGGGAATGAGCCCTGCGCCTGCGCGCACCCCGCCACCCCCTCCAG USF NFkB B SP1 GCCGGAGTCCACGCCCACTTGGGGGAGGAGCCCCGCAGCCTCCACCTACAGGGGCGTCCCCT C AP-1/NFkB SP1 AAGGGGACGGAGGCCGCATGGGCCGCCGAGCCGGGAAATCTCCGCCCCCAGCTGGAGCGGCT A +1 D GTGCGGGCTGCGTAGCGGTGCTGGGTCGGGCCGACGTGCCACCCACCCGGAGCCGGTGAGTG +90 CAGCCGCCCGCCCTCCGGTAGATCTGCGGCCTGGCGGAGAAGTCGGGAGGGGACAGGAAGGG AGGGCGGGCCCCGGGCCCTCCTCCGTCTCAGCCGCCTGCGGAGGTGCTGCCCACGCCTGGAGG CCCCCACTGACCCTCAGACCCGCGTCTGCGCCCCTCTCCCCGCACCCCGAGGCAGAGTTGGGA AAGCAGTGGTCTTAGACCCCCCACCTCGGGCACTCGGAAGAGAACGGCGGAGACAACCCCTC CTCTTCCCTGGCTGGCGCAGCGCCAGCCTCGAGCTCCTCGGTAGCCCCCGGGCAGGGAGGGCC GGAGGGTGGGCGCGGCATCTTCAGCGACTCTTCGAAGTCCCTTCCGCGTCTCATCTTTCAAGG CTGTTGCAGAGGCGGCTTGCTTCCCACCTGTCCATCTCCATAAAAATCCCTAAACGAAACATG AP-1/CREB CCCACGTGTCCGGAGATTTTCAGGACTTGGTGCATTTCAGATGAAGGCTTTTCCAGAAGCTTCC CCGTAGAAGAGGATCAGGCATCCAACTGGTTAAGGTCAGCAGCGTTTGGCACGTCTCCTTCCA GCCTGGCGGTTTTGTCAGGATTCCCTGGGGAGTGTCTGGAAAGCCTGATGAGGGGAAATAGTA CATCTCAGCGAATCGGCACCAGCGAGTGTAAGATGCGCGTTATTGAATGTG (+710) 60 Figure 11: The DNA sequence, encompassing the pSTEP1 fragment, that includes the NEIL2 gene sequence flanking 5’ upstream and 3’ downstream of the identified transcriptional start site (+1). Putative consensus sequences for binding to regulatory factors are AP-1 (highlighted), CREB (italicized), NFkappaB (underlined). Rectangles indicate locations of single base pair mutations: A = rs8191518, B = NFkappaB-1 (-104 G to C), C = SNP ss74800505, D = SNP ss74800504. This yielded three variations of the wild type p1200C plasmid with single mutations (Table VI). A second mutation was then introduced into each of the p1200C and pSTEP1 plasmids that already had the ss74800505 mutation, using the same technique and respective oligos (Table V) as described above, and using the p1200C and pSTEP1 + ss74800505 as the template for PCR amplification. This created a total of six variations of each wild type plasmid (Table VI). The accuracy of the second base change was verified by direct sequencing as well. Cell culture, transfection and glucose oxidase treatment MRC-5 cells were cultured as previously described in Chapter 2, Materials and Methods. Twenty-four hours prior to transfection, cells were harvested from the flasks, counted and seeded in 6-well plates at a concentration of 1.6x105 cells/well. MRC-5 cells were transfected with the constructs as described in Chapter 2, Materials and Methods using a mixture of 1.0 µg luciferase and 0.5 µg Renilla DNA/well at a Lipofectamine (Invitrogen, Inc.) concentration of 6 of Lipofectamine/µg plasmid DNA. Transfection efficiency was tested using the Dual Luciferase Reporter System (Promega, Inc.). Luciferase reporter gene expression was detected as described in Chapter 2, Materials and Methods using the Dual Luciferase Reporter System (Promega, Inc.). Luminescence was measured in relative light units (RLUs) per L. Each experiment was repeated at least three times. In addition, MRC-5 cells transfected with either the p1200C wild type (bp -206 to +90) or p1200C (-104 GtoC) luciferase construct were treated with 100 ng/ml the cellular oxidant GO for one hour as described in Chapter 2.(GO; Roche, Inc. Basel, Switzerland). 61 Statistical analysis. The Student's t-test or ANOVA was used, when appropriate, to compare the mean luciferase and Renilla expression between different plasmid constructs. All experiments were repeated at least three times, in duplicate. All data are presented as means ± standard error (SE). A two-sided P<0.05 was considered statistically significant. All statistical analyses were performed using NCSS 2004 (Kaysville, UT) or EXCEL (Microsoft, Inc.) software. Results Table VI shows the combinations of mutations engineered into each plasmid using site directed mutagenesis. Point mutations corresponding to reported SNPs as well as in a robust transcriptional element binding site were introduced into each plasmid. The previously reported location of each SNP was used to dictate the sitedirected mutagenesis for plasmids containing SNPs rs8191518, ss74800504 and ss74800505 (dbSNP, NCBI). The TESS software was then used to analyze the NEIL2 promoter region in each of the plasmids for putative transcription-factor binding motifs (TESS, University of Pennsylvania, PA). The results of this analysis showed several motifs, including an overlapping site for the binding of NF-kappaB and Sp-1 trans-acting factors at the -104 site (Figure 11. This binding site was chosen as a target because both of these trans-acting factors are activated by oxidative stress (Muller and Gawlik, 1997; Schreck et al., 1991). The weighted matrix option of the TESS software was used to show that a G to C mutation at -104 bp 5’ from the transcriptional start site would confer a sequence change that would disrupt the binding of both the NF-kappaB and Sp-1 proteins, thus one mutation would test if one of these factors played a role in the regulation of the NEIL2 transcript (Figure 11. The data indicate that insertion of single base changes corresponding to SNP ss74800505, ss74800504 and rs8191518 resulted in no significant change in expression compared to the wildtype pSTEP1 plasmid (Figure 12) 62 * Relative light units 6 * 5 4 3 2 1 0 pSTEP1 wt pSTEP1 + pSTEP1 + ss74800505 ss74800504 pSTEP1 + rs8191518 pSTEP1 + pSTEP1 + pSTEP1 + ss74800505 ss74800505 ss74800505 + + rs8191518 + (-104 GtoC) ss74800504 Figure 12: Promoter activity from pSTEP1 constructs containing site-directed mutations (determined in relative light units as described in Materials and Methods) * P < 0.05. Similarly, SNPs ss74800505, and ss74800504, and a mutation at the NF-kappaB site (104 GtoC) resulted in no significant change in expression compared to the levels of expression from the wild type p1200C plasmid (Figure 13. The SNP ss74800504 moderately induced expression levels above the level observed with the wild type p1200C plasmid (P=0.054; Figure 13. 63 Relative light units 60 50 40 30 20 * * 10 0 p1200Cwt p1200C + ss74800505 p1200C + ss74800504 p1200C + (p1200C + p1200C + p1200C + 104 GtoC) ss74800505 + ss74800505 + ss74800505 + ss74800504 rs8191518 (-104 GtoC) Figure 13: Promoter activity from p1200C constructs containing site directed mutations (determined in relative light units as described in Materials and Methods) * P < 0.05. To test how combinations of base changes would affect NEIL2 expression, a set of point mutations were engineered into each plasmid (Figure 11 Table V). Insertion of mutations corresponding to the combination of SNP ss74800505 and SNP rs8191518 into pSTEP1 resulted in a 2-fold increase in expression over wild type (P=0.03), and a 2.6fold increase over SNP ss74800505 alone (P=0.02; Figure 12. Insertion of mutations corresponding to the SNP ss74800505 and the -104 GtoC mutation in NF-kappaB/Sp-1 resulted in a 3-fold increase in expression over that observed with the wild type (P<0.01) and a 3.7- fold increase over that observed with SNP ss74800505 alone (P<0.01; Figure 12. In contrast, insertion of both SNPs ss74800505 together with rs8191518 into the p1200C plasmid resulted in a 53% reduction in expression relative to the wild type form (P<0.01), and a 69% reduction relative to the ss74800505 SNP alone (P<0.01; Figure 13. Insertion of the ss74800505 SNP, together with the mutation in NF-kappaB (-104 GtoC), resulted in a 47% decrease in expression relative to the level observed with the wildtype (P=0.02) and a 53% decrease in expression relative to ss74800505 alone (P<0.01). To test the response of our NEIL2 promoter constructs to excessive ROS, we exposed transfected cells to GO. Glucose oxidase treatment of cells containing the 64 p1200C wild type construct resulted in a 46% decrease in expression relative to untreated transfected cells (P<0.01; Figure 14. No change in expression was observed between untreated cells and treated cells transfected with the p1200C plasmid containing the -104 GtoC mutation in the NF-kappaB/Sp-1 binding site (Figure 14. * 25 20 15 10 5 0 p1200Cwt p1200Cwt + GO p1200C + (-104 GtoC) p1200C + (-104 GtoC) + GO Figure 14 Promoter activity for p1200C wild type and p1200C + mutation in NFkappaB site (104 GtoC) with and without 1 hr GO treatments (determined in relative light units as described in Materials and Methods) * P < 0.05 DISCUSSION The goal of this study was to test the hypothesis that single base mutations in the NEIL2-promoter region affect gene expression, in vitro. To our knowledge, this is the first study to evaluate the mechanistic role of NEIL2 SNPs or to test the effect of a mutation in the putative NF-kappaB and Sp-1 site (-104 GtoC) on gene transcription. In previous studies presented in Chapter 1 of this dissertation, we showed that SNP rs8191518 was primarily present in high expressers of NEIL2 and that SNP ss74800505 was associated with an increase in mutagen sensitivity, indicating a potential relationship between these SNPs and disease risk (Table II, III). Furthermore, the data presented in Chapter 1 of this dissertation showed that SNPs ss74800505 and rs8191518 were present in a frequency of 0.33 in the population, and with a linkage disequilibrium 65 of D’=0.73 (see Chapter 1). Taken together, these data suggests that the presence of these two SNPs, either independently or together, could alter NEIL2 gene expression and, thus, could affect disease risk. Therefore, investigations into the mechanistic role of the co-occurrence of SNPs ss74800505 and rs8191518 were conducted. In the current study, an increase in NEIL2-driven expression from plasmid pSTEP1 was observed, when both SNP ss74800505 and SNP rs8191518 were present. These results support our in vivo findings that showed a higher frequency of SNP rs8191518 in high expressers (Table II). Interestingly, in silico analyses of these base changes did not predict that there would be a direct disruption of transcription factor binding. However, SNP ss74800505 lies near an NF-kappaB/AP-1 motif (Figure 11, thus a change at this location could potentially enhance the activation of transcription from an NF-kappaB or AP-1 trans-acting protein in this construct. Such an affect could lead to tighter binding or increased recruitment of these positive transcription factors. Alternatively, this might be an additive positive effect of both SNPs, resulting in increased gene expression. Another possible explanation of our findings is that these base changes could be affecting transcriptional regulation further down-stream of the SNPs. Our previous studies in Chapter 2 of this dissertation have shown that the pSTEP1 plasmid contains a negative regulatory region (+49 to +710) that suppresses the adjacent, positive (-206to +90) regulatory region (Figures 7-9 Although the silencing motif in the NEIL2 promoter (+49 to +710) has not been completely characterized, we hypothesize that mutations upstream of this region could change the architecture of the DNA, so that the respective silencing factors cannot recognize the binding motif. This hypothesis is consistent with results from several studies that have shown that transcriptional regulation is, in part, dependent on the flexibility of the DNA, and that single base changes near the gene transcription start site can change the ability of the DNA to bend to accommodate the transcriptional protein complex (Breit et al., 2008; Strahs et al., 2003; Stepanek et al., 2007; Huet et al., 2005). An example of this is the TATA element, present in many eukaryotic promoters, and which is recognized by the TATA box binding protein (TBP) 66 through its intrinsic shape (Strahs et al., 2003). Single base changes in this element promote less favorable geometries and result in decreased transcriptional activity (Strahs et al., 2003). One hypothesis that is worthwhile pursuing in the future is that the base changes introduced into the pSTEP1 construct have altered transcription in a similar manner by disrupting the silencing mechanism that lies downstream of the start site. This would result in an impedance of the silencing properties, and therefore, an increase in gene expression. The p1200C plasmid contains a strong transcription-enhancing region (-206 to +90) isolated from the NEIL2 promoter (Figure 9. Insertion of both SNPs ss74800505 and SNP rs8191518 into this region resulted in a 2-fold decrease in expression (Figure 13. One possible explanation for this decreased expression is that two nucleic acid base changes resulted in architectural alterations in the DNA such that binding of transcriptional proteins was limited. This result is in contrast to the observations of the single base change effects in the larger pSTEP1 fragment. This result is not unexpected considering the p1200C plasmid contained only a portion of the length and regulatory motifs as that of the pSTEP1 plasmid. Thus, single base changes in the p1200C fragment could result in dramatically different architectural changes than that of the pSTEP1 fragment and result in the binding of different transcriptional regulating proteins. An in silico search for putative DNA binding motifs identified several strong cisacting elements, including an over-lapping NF-kappaB and SP-1 site (-104; Figure 1). These sites are interesting because they bind ROS-responsive transcription factors (Schoonbroodt and Piette, 2000). Thus, one could propose that single base changes at this location could change the transcription factor binding affinity to this site and result in an alteration of gene transcription, particularly in the presence of oxidative stress. Consistent with this hypothesis, the single mutation at the NF-kappaB/Sp-1 site in combination with SNP ss74800505, increased expression significantly compared to levels observed when the SNP ss74800505 was introduced alone into the pSTEP1 plasmid (Figure 12. A mutation at this site also conferred an increase in expression in combination with SNP ss74800505 in the p1200C construct. These findings suggest and 67 integral role for SNP ss74800505 in combination with the NF-kappaB/Sp-1 in regulation of NEIL2 promoter activity. In our studies described in Chapter 2, we have shown that promoter-driven expression of the 1200C construct is decreased after treatment with GO (Figure 10 In this study, the mutation at the NF-kappaB/Sp-1 site (-104 GtoC) was found to abolished this response. These results indicate that the NF-kappaB/Sp-1 motif at bp -104 plays a critical role in regulating NEIL2 gene expression in response to oxidative stress. The mechanism by which NF-kappaB is activated in the present of oxidative stress, and how it promotes gene expression in response to increased cellular ROS and inflammatory events is well known (Schoonbroodt and Piette, 2000). Breifly, NF-kappaB is sequestered in the cellular cytoplasm by binding to I-kappaB (Schoonbroodt and Piette, 2000). ROS induces a signaling cascade that results in the release of NF-kappaB from IkappaB (Schoonbroodt and Piette, 2000). NF-kappaB then moves into the nucleus to act as a transcription factor (Schoonbroodt and Piette, 2000). Several studies have shown that NF-kappaB binds and induces gene transcription (Schoonbroodt and Piette, 2000). In contrast to the enhancement of expression, other studies have shown that oxidative stress-mediated activation of NF-kappaB repressed expression of several genes, including human 25-hydroxyvitamine D3 1-hydrolase, estrogen receptor, and 1-adrenoceptor (Zhang et al., 2007; Van Laere et al., 2007, and Ebert et al., 2008). These studies have shown that ROS activates the translocation of NF-kappaB protein into the nucleus, where it binds to promoter regions of specific genes to repress expression (Zhang et al., 2007; Van Laere et al., 2007, and Ebert et al., 2008). We hypothesize that NF-kappaB down regulates the NEIL2 promoter fragment in the p1200C wild type construct by binding to and repressing transcription because this response was abolished with the point mutation in the NF-kappaB site (-104 GtoC) and no down-regulation of gene expression was observed in the presence of GO. Taken together, the studies described in this chapter show that combinations of single base changes in the NEIL2 promoter region alter gene expression in vitro. Also, the findings indicate that NF-kappaB signaling plays a critical role in NEIL2 expression 68 in the presence of oxidative stress. These results provide suitable mechanistic explanation for the observed inter-individual differences in NEIL2 expression observed in population studies described in Chapter 1 of this dissertation (Figure 2). Results from these studies open several avenues for future investigations, including further characterization of the diverse nature of the NEIL2 promoter and larger population studies to help understand the role of NEIL2 promoter SNPs and disease risk. 69 CHAPTER 5: CONCLUSIONS We found that transcription levels of the NEIL2 gene displayed a wide interindividual variation in our population of subjects and that this variation could depend on the presence of SNPs in the promoter region. This implies a wide range of interindividual variability in NEIL2-mediated DNA repair. Results presented in Table II of Chapter 1, illustrate that several SNPs were associated with a high expresser phenotype, while others were associated with a low expresser phenotype. This indicates that NEIL2 expression is influenced by polymorphic variations in the promoter region of the gene. The expression level was associated with ethnicity (Figure 3). Ethnicity is well known to be an important determining factor for susceptibility to diseases associated with oxidative stress, such as heart disease and diabetes, for example (Davidson et al., 2007; Gaskin et al, 2001). Thus, ethnic groups with higher NEIL2 transcript levels could be at lower risk for disease, relative to other groups. Our mechanistic studies have shown that NEIL2 promoter SNPs ss74800505 and ss74800504 were associated with increased mutagen sensitivity (Table III). As such, these studies identify critical, heritable factors in the promoter region of the NEIL2 gene that could be considered, in the future, to project an individual’s response to mutagenic insults. Results from the mechanistic studies show that the combination of SNP rs8191518 and the newly identified SNP ss74800505 significantly changes NEIL2 promoter-driven expression in vitro (Figures 11=2nd 13. These observations open the door for several hypothesis that could be tested. One hypothesis is that polymorphic regions are modifying the binding affinity of transcriptional proteins along the NEIL2 promoter region. In vitro results of luciferase constructs containing these polymorphic sites support this hypothesis, by showing that constructs containing these SNPs have altered expression levels. Furthermore, we have shown in Figure 14 that modifying an 70 NF-kappaB/Sp-1 binding motif, lying upstream of the transcriptional start site influences NEIL2 expression when under excessive cellular oxidative stress. The identification of an NF-kappaB/Sp-1 binding motif as a potential important regulatory region is significant because these proteins are influenced by intra-cellular ROS. In most studies, ROS induce the expression of genes through the activation of transcription factors, but there are exceptions in the literature that demonstrate decreased transcription factor binding in the presence of ROS (Reviewed by Allen and Tresini, 2000). For example, ROS reduced the DNA binding affinity of the CRE-binding protein (CREB) and hypoxia-inducable factor 1 (HIF1) transcription factors. Furthermore, ROS are also associated with reduced expression of several genes, including c-fos, IL-2, CYP1A1, and CYP1A2 (Allen and Tresini, 2000). ROS also influence gene transcription through the activation of transcriptional regulatory pathway proteins, such as NF-kappaB and AP-1 (Allen and Tresini, 2000). This includes the activation of BER enzymes, such as APE1 and NEIL1 (Allen and Tresini, 2000, Das et al., 2005). ROS can also influence proteins in a post-transcriptional manner. Das et al. (2007) showed that NEIL2 glycosylase activity was enhanced in the presence of oxidative stress through enhanced protein-protein binding of NEIL2 to the Y-box-binding protein-1 (YB-1). With enhanced activity, NEIL2 could play a larger role in BER. This also suggests that the BER pathway is a fluid mechanism that reacts to external, damaging influences, such as oxidative stress. For example, ROS are known to enhance the activation of the DNA repair-associated protein, p53 as well as enzymes in several transcriptional regulatory pathways (Reviewed by Allen and Tresini, 2000). The current project shows that NEIL2 is regulated at the transcriptional level, but in contrast to the report of reported enhanced activation of this glycosylase in the presence of oxidative stress (Das et al., 2007), NEIL2 gene transcription is repressed in the presence of oxidative stress produced by GO. Sequence analysis shows that this response could be influenced by the presence of SNPs in the promoter region of this gene. Thus, there could be a genetic component influencing the down regulation of NEIL2 transcription in response to oxidative stress. 71 One possible explanation for why NEIL2 activity would be enhanced in the presence of ROS, as reported by Das et al., (2007), while transcription is repressed, is that the initial response to oxidative stress may be through the activation of the NEIL2 enzyme that is already present, followed by conservation of the transcriptional regulatory machinery for the activation of other proteins. Because NEIL2 glycosylase/AP lyase activity is the first step in BER, it would be logical for it to be quickly activated to initiate the repair process. That leaves time and cellular resources (such as transcriptional regulatory factors) for the transcription and translation of other BER enzymes that are responsible for later repair steps. Thus, transcriptional conservation of NEIL2 could be part of the diversion of important transcriptional factors to the up-regulation of genes that are important for steps farther down the enzymatic pathway. In this way, the cell could use stored resources to efficiently initiate damage repair. In vitro studies presented in Chapter 2 of this dissertation showed that the NEIL2 expression level decreased and then rebound after twelve hours of exposure to GO. This may illustrate an initial conservation of transcription by repression, followed by a release of this repression once the cellular stores of NEIL2 protein were depleted due to oxidative stress. The results of these studies also suggest that NEIL2 expression patterns could be related to the global coordination of cellular DNA repair that is needed to ensure genetic stability. ROS and ionizing radicals create highly diverse types of single-strand breaks that are repaired by the coordinated action of proteins in the BER pathway (Dianov and Parsons, 2007). The complexity of these lesions could require unique subsets of BER enzymes to work together. However, uncertainty persists about which subset of proteins repair which lesion(s) (Dianov and Parsons, 2007). Because of the novelty of the NEIL2 protein, little is known about how it interacts with other glycosylases, or how its protein activity or gene expression could be affected by the type of lesion formed. Furthermore, DNA glycosylases are known to interact with other non-BER pathways and this relationship has an increasing important role in directing repair of DNA lesions (Kovtun and McMurray, 2007). DNA glycosylases have been reported to interact with mismatch repair (MMR) as well as nucleotide excision repair pathways (Kovtun and McMurray, 72 2007). For example, when the highly mutagenic lesion 8-oxoG is paired with a cytosine, it is removed by the BER-specific glycosylase OGG1 (Kovtun and McMurray, 2007). However, if it is paired with an adenine, the adenine is first removed by the MMRspecific enzyme, MYH (Kovtun and McMurray, 2007). This leaves the 8-oxoG paired with a cytosine, the base-pair substrate for OGG1 (Kovtun and McMurray, 2007). These observations suggest that NEIL2 could be involved in other DNA repair pathways. As such, the observed decrease in expression of this gene in the presence of GO could be due to cellular conservation and the balancing of available proteins to optimize DNA repair under excessive oxidative stress. These logical explanations could be tested in future studies. The information gained from this project could have substantial implications for basic research as well as clinical applications. Our mechanistic studies paved the way for future projects that could use our NEIL2 plasmid constructs as ‘molecular tools’ to examine the effects of other environmental factors, such as UV light, on NEIL2 expression patterns in vitro. Such experimental studies would provide a better understanding of the role of NEIL2 expression in disease prevention, particularly in the presence of environmental stresses. This project also has significant practical clinical implications in the area of cancer treatment. As discussed in the introduction, disruption of the BER pathway has been considered as a supplementary treatment to enhance chemotherapeutic efficacy. Understanding the factors that influence the regulation of this gene, could help enhance cancer treatment by disrupting the BER pathway by altering NEIL2 transcription. Furthermore, we found that two promoter SNPs are in linkage disequilibrium and that the co-occurrence of these SNPs alters NEIL2 transcription in vitro. This implies that certain haplotypes in the NEIL2 promoter region could predispose certain individuals for developing adverse health affects from ROS generating agents. Alternatively, it could be used it identify people at low risk for developing genetic-damage based disease as complications of conditions associated with an increase in ROS, such as arthrosclerosis or diabetes (American Lung Society, 2008). 73 Future studies Delineation of NEIL2 regulatory motifs We have shown that the NEIL2 gene has several distinctive regulatory regions. Additional studies are needed to further delineate the exact sequences responsible for induction and suppression of NEIL2 expression. Such studies could include DNA footprint analysis to identify transcription factor proteins that bind directly to the sequences within the positive (-206 to +90) and negative (+49 to +710) regions. Such investigations have been informative for the promoter element of the NEIL1 gene (Das et al., 2005). These assays could be performed under the conditions of low and high oxidative stress to define the influence of these conditions on the binding of the factors that drive NEIL2 expression. Haplotype analysis of the NEIL2 promoter region Our laboratory has isolated and frozen, in accordance with a previously established protocol, samples of DNA from human subjects. This has created a bank of genomic DNA from over 700 individuals. We also have extensive demographic information on these individuals, including age, medications used, and smoking status, and other lifestyle factors, as well as data regarding the sensitivity of the lymphocytes from these individuals to certain mutagens. Because of the availability of these DNA samples, new sequencing studies could be designed for specific questions. For example, sequencing a larger portion of the NEIL2 promoter region would allow one to create haplotype tags that could be associated with mutagen sensitivity and possible disease risk. Such studies have been completed for several genes, including genes that code for the repair enzyme, XRCC1, and ERCC2 (Affatato et al., 2004; Hao et al., 2004; Ng et al., 2008; Pakakasama et al., 2007). 74 Transfection of NEIL2 promoter constructs into other cell types The constructs created in this study should be tested in other cell lines that have been associated with exposure to ROS in vivo. For example, muscle cells are known to have very high metabolic activity and would be more likely to have a higher internal level of ROS. Northern blot analysis by Hazra et al., (2000) showed muscle tissue had the highest amount of NEIL2 expression, relative to several other tissues. It would be worthwhile to evaluate how muscle cells regulate NEIL2 expression differently than other tissues which have a lower expression. If one could elucidate the factors that bind and enhance NEIL2 expression, then the next step is to identify how modifications in the binding motif for these factors affect gene transcription. 75 APPENDIX Table VII: Distribution of NEIL2 and XPD expression in the study population. ID S-310 S-311 S-385 S-294 S-271 S-305 S-315 S-295 S-246 S-418 S-475 S-407 S-379 S-360 S-404 S-155 S-417 S-391 S-218 S-296 S-558 S-297 S-617 S-301 S-421 S-448 S-346 S-648 S-344 S-200 S-291 S-357 S-317 S-309 S-347 S-832 S-402 S-711 S-744 S-469 S-447 S-863 S-489 S-427 S-638 S-767 S-520 S-490 S-461 S-838 S-444 S-170 S-375 S-669 S-470 S-401 S-422 S-728 S-434 S-864 S-672 S-705 S-677 S-451 S-458 S-843 S-268 XPD expression 11054 14864 12024 21785 15098 21244 6682 12911 9945 11823 14666 13715 15206 16658 13742 17146 10941 7906 12552 37360 9739 9626 9511 14856 25981 9484 10033 11250 19178 13765 13633 18791 15182 17554 11974 22391 14588 14281 13267 14453 21251 15927 6909 15705 24996 24309 Smoking status NEIL2 expression non-smoker non-smoker smoker smoker ex-smoker ex-smoker ex-smoker non-smoker Smoker ex-smoker non-smoker non-smoker non-smoker non-smoker smoker ex-smoker ex-smoker smoker smoker smoker ex-smoker smoker non-smoker non-smoker ex-smoker smoker non-smoker smoker ex-smoker smoker non-smoker ex-smoker non-smoker non-smoker non-smoker smoker ex-smoker ex-smoker non-smoker non-smoker non-smoker smoker smoker non-smoker ex-smoker non-smoker non-smoker non-smoker non-smoker non-smoker ex-smoker Smoker non-smoker ex-smoker ex-smoker non-smoker ex-smoker non-smoker non-smoker smoker non-smoker non-smoker non-smoker smoker non-smoker non-smoker non-smoker 429.13 455 560.29 574.38 630.65 633.02 644.23 847.97 882.32 893.69 951.96 1015.67 1022.78 1042.01 1046.51 1051.44 1054.99 1082.43 1087.51 1100.4 1119.69 1126.13 1127.5 1140.25 1165.02 1186.61 1188.54 1197.9 1199.9 1270.26 1337.38 1368.24 1378.55 1401.2 1433.06 1448 1465.93 1477.96 1509.17 1519.94 1520.41 1573 1592.05 1595.37 1616.12 1628.91 1668.86 1696.37 1753.97 1755 1772.01 1772.16 1866.76 1886.39 1898.15 1949.12 1968.27 1979.29 1997.88 2004 2031.86 2035.78 2105.63 2164.01 2168.62 2172 2184.07 ID S-199 S-674 S-367 S-387 S-322 S-725 S-527 S-438 S-693 S-670 S-365 S-465 S-801 S-352 S-555 S-409 S-594 S-800 S-733 S-853 S-437 S-307 S-509 S-720 S-668 S-579 S-736 S-828 S-140 S-580 S-856 S-425 S-399 S-543 S-703 S-574 S-782 S-618 S-664 S-454 S-860 S-865 S-453 S-521 S-554 S-545 S-126 S-785 S-234 S-764 S-147 S-467 S-338 S-715 S-396 S-612 S-667 S-436 S-629 S-587 S-538 S-150 76 XPD expression 13946 14910 19630 27341 19190 11913 40107 29465 14179 12988 18584 19645 16536 37897 34008 15369 6533 10912 8661 14002 7152 25185 10754 15325 11668 13466 19376 12882 27654 18035 14918 22488 Smoking status NEIL2 expression Smoker non-smoker non-smoker smoker smoker ex-smoker non-smoker non-smoker non-smoker smoker ex-smoker ex-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker smoker smoker ex-smoker ex-smoker ex-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker non-smoker smoker non-smoker non-smoker ex-smoker smoker smoker ex-smoker non-smoker non-smoker smoker smoker smoker smoker smoker non-smoker smoker non-smoker smoker non-smoker non-smoker smoker non-smoker non-smoker non-smoker smoker non-smoker 2185.41 2210.74 2231.35 2232.94 2280.5 2285.16 2318.81 2327.67 2327.99 2328.38 2346.76 2357.81 2383 2390.75 2489.53 2512.53 2519.25 2537 2580.66 2584 2584.71 2619.86 2654.1 2672.27 2699.32 2715.73 2795.85 2823 2827.18 2959 2973 3027.81 3031.7 3153.87 3234.27 3259.82 3346.79 3351 3383 3396.3 3564 3580 3591.34 3711.61 3857.51 3863.21 4139.71 6971.99 7179.33 7964.3 10109.11 10982.44 11771.25 11834.52 14703.24 14911.98 17037.57 17466.16 18847.63 19835.65 20019.61 27183.42 Table VIII: Distribution of ethnicities, high or low NEIL2 and accumulated chromosome breaks in study population. ID Ethnicity S-310 S-311 S-385 S-294 S-271 S-305 S-315 S-295 S-246 S-418 S-475 S-407 S-379 S-360 S-404 S-155 S-417 S-391 S-218 S-296 S-558 S-297 S-617 S-301 S-421 S-448 S-346 S-648 S-344 S-200 S-291 S-357 S-317 S-309 S-347 S-832 S-402 S-711 S-744 S-469 S-447 S-863 S-489 S-427 S-638 S-767 S-520 S-490 S-461 S-838 S-444 S-170 S-375 S-669 S-470 S-401 S-422 S-728 S-434 S-864 S-672 S-705 S-677 S-451 S-458 S-843 S-268 non-Hispanic White non-Hispanic White African American African American non-Hispanic White non-Hispanic White Hispanic non-Hispanic White non-Hispanic White non-Hispanic White African American African American non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White Asian African American non-Hispanic White Hispanic non-Hispanic White African American non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White Asian Hispanic non-Hispanic White African American non-Hispanic White African American non-Hispanic White Asian non-Hispanic White Hispanic Asian non-Hispanic White African American Asian non-Hispanic White Asian non-Hispanic White Hispanic non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White Asian non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White Hispanic non-Hispanic White non-Hispanic White non-Hispanic White High or low NEIL2 expression low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 low NEIL2 expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression expression Accumulated breaks 48 hr after stimulation 1 1 0 3 3 0 0 4 1 1 2 4 0 1 1 1 0 4 1 1 7 0 1 0 2 2 5 9 4 3 2 77 ID Ethnicity S-199 S-674 S-367 S-387 S-322 S-725 S-527 S-438 S-693 S-670 S-365 S-465 S-801 S-352 S-555 S-409 S-594 S-800 S-733 S-853 S-437 S-307 S-509 S-720 S-668 S-579 S-736 S-828 S-140 S-580 S-856 S-425 S-399 S-543 S-703 S-574 S-782 S-618 S-664 S-454 S-860 S-865 S-453 S-521 S-554 S-545 S-126 S-785 S-234 S-764 S-147 S-467 S-338 S-715 S-396 S-612 S-667 S-436 S-629 S-587 S-538 S-150 non-Hispanic White non-Hispanic White non-Hispanic White American Indian non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White African American African American non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White Asian non-Hispanic White African American non-Hispanic White non-Hispanic White non-Hispanic White African American African American Hispanic non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White Asian Hispanic Hispanic non-Hispanic White African American non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White African American Asian non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White Hispanic non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White non-Hispanic White Accumulated breaks 48 hr High or low after NEIL2 expression stimulation low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression low NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression high NEIL2 expression 3 0 0 5 2 5 -1 1 4 5 1 3 5 3 3 1 2 6 3 1 4 2 0 1 2 1 1 3 3 2 3 Table IX: Mean luciferase expression in NEIL2 promoter clones. Plasmid P1200 pSTEP3 pSTEP2 pSTEP1 p1200C p1200B p1200A P1200 Mean RLU's/g total protein 241.36 33.78 48.53 43.57 219.82 63.10 32.23 241.36 SE 39.64 5.69 1.72 2.71 25.87 8.54 3.89 39.64 Table X: Mean luciferase expression in pSV-40-luciferase clones Plasmid Mean RLU's/g total protein SE 5473.63 568.24 516.17 64.70 pSV40 pSV40Step3 Table XI: Mean luciferase expression in p1200C clones exposed to GO. Plasmid Time p1200C P1200c p1200C p1200C pNEIL1 pNEIL1 pNEIL1 pNEIL1 0hr 1hr 6hr 12hr 0hr 1hr 6hr 12hr Mean RLU’s SE 3.27 2.29 1.98 2.83 1.13 1.25 0.49 0.29 0.25 0.24 0.14 0.23 0.07 0.12 0.11 0.10 78 Table XII: Mean luciferase expression in pSTEP1 clones Plasmid with mutation Mean RLU's SE pSTEP1 wt pSTEP1 + ss74800505 pSTEP1 + ss74800504 pSTEP1 + rs8191518 pSTEP1 + ss74800505 + ss74800504 pSTEP1 + ss74800505 + rs8191518 pSTEP1 + ss74800505 + (-104 GtoC) 1.71 1.38 2.10 1.33 1.95 3.70 5.15 0.34 0.45 0.27 0.28 0.31 0.47 0.30 Table XIII: Mean luciferase expression in p1200C clones Plasmid with mutation RLU's SE p1200Cwt p1200C + ss74800505 p1200C + ss74800504 p1200C + (-104 GtoC) p1200C + ss74800505 + ss74800504 p1200C + ss74800505 + rs8191518 p1200C + ss74800505 + (-104 GtoC) 16.37 18.55 38.74 14.84 12.90 7.80 8.75 2.62 3.01 9.90 1.20 1.74 0.56 0.58 Table XIV: Mean luciferase expression in p1200C clone with mutation in the NFkB/Sp1 site. Plasmid with mutation RLU's SE 1200C wt 1200C wt + GO 1200C wt + (-104 GtoC) 1200C wt + (-104 GtoC) + GO 16.37 9.02 14.84 16.87 2.62 0.62 1.20 1.64 79 REFERENCES Affatato AA, Wolfe KJ, Lopez MS, Hallberg C, Ammenheuser MM, bdel-Rahman SZ.Effect of XPD/ERCC2 polymorphisms on chromosome aberration frequencies in smokers and on sensitivity to the mutagenic tobacco-specific nitrosamine NNK. Environ Mol Mutagen 2004;44:65-73. Alberts B. Molecular biology of the cell, 2nd ed ed. New York: Garland Pub. Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med 2000;28:463-99. Abdel-Rahman SZ, Soliman AS, Bondy ML et al. Inheritance of the 194Trp and the 399Gln variant alleles of the DNA repair gene XRCC1 are associated with increased risk of early-onset colorectal carcinoma in Egypt. Cancer Lett 2000;159:79-86. Berwick M, Vineis P. Markers of DNA repair and susceptibility to cancer in humans: an epidemiologic review. J Natl Cancer Inst 2000;92:874-97. Blasiak J, Arabski M, Krupa R et al. Basal, oxidative and alkylative DNA damage, DNA repair efficacy and mutagen sensitivity in breast cancer. Mutat Res 2004;554:139-48. Bonassi S, Abbondandolo A, Camurri L et al. Are chromosome aberrations in circulating lymphocytes predictive of future cancer onset in humans? Preliminary results of an Italian cohort study. Cancer Genet Cytogenet 1995;79:133-35. Bonassi S, Neri M, Puntoni R. Validation of biomarkers as early predictors of disease. Mutat Res 2001;480-481:349-58. Bowler RP, Barnes PJ, Crapo JD. The role of oxidative stress in chronic obstructive pulmonary disease. COPD 2004;1:255-77. Breit JF, ult-Ziel K, Al-Mehdi AB, Gillespie MN. Nuclear protein-induced bending and flexing of the hypoxic response element of the rat vascular endothelial growth factor promoter. FASEB J 2008;22:19-29. 80 Broderick P, Bagratuni T, Vijayakrishnan J, Lubbe S, Chandler I, Houlston RS. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1 genes in familial colorectal cancer predisposition. BMC Cancer 2006;6:243. Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 2000;25:169-93. Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT. Mapping determinants of human gene expression by regional and genome-wide association. Nature 2005;437:1365-69. Chuang CH, Hu ML. Synergistic DNA damage and lipid peroxidation in cultured human white blood cells exposed to 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone and ultraviolet A. Environ Mol Mutagen 2006;47:73-81. Chuang CH, Hu ML. Synergistic DNA damage and lipid peroxidation in cultured human white blood cells exposed to 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone and ultraviolet A. Environ Mol Mutagen 2006;47:73-81. Cloutier JF, Drouin R, Weinfeld M, O'Connor TR, Castonguay A. Characterization and mapping of DNA damage induced by reactive metabolites of 4-(methylnitrosamino)-1(3-pyridyl)-1-butanone (NNK) at nucleotide resolution in human genomic DNA. J Mol Biol 2001;313:539-57. de W, Jr., Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol 1987;7:725-37. DeLuca M, Wannlund J, McElroy WD. Factors affecting the kinetics of light emission from crude and purified firefly luciferase. Anal Biochem 1979;95:194-98. Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem 2003;278:49679-84. Egyhazi S, Ma S, Smoczynski K, Hansson J, Platz A, Ringborg U. Novel O6methylguanine-DNA methyltransferase SNPs: a frequency comparison of patients with familial melanoma and healthy individuals in Sweden. Hum Mutat 2002;20:408-9. 81 El-Zein R, Abdel-Rahman SZ, Conforti-Froes N, Alpard SK, Zwischenberger JB. Chromosome aberrations as a predictor of clinical outcome for smoking associated lung cancer. Cancer Lett 2000;158:65-71. El-Zein RA, Hay MJ, Lopez MS et al. Response to comments on 'Cytogenetic effects in children treated with methylphenidate' by El-Zein et al. Cancer Lett 2006;231:146-48. Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 2004;567:1-61. Faniello MC, Fregola A, Nistico A et al. Detection and functional analysis of an SNP in the promoter of the human ferritin H gene that modulates the gene expression. Gene 2006;377:1-5. Flavell RA, Sabo DL, Bandle EF, Weissmann C. Site-directed mutagenesis: generation of an extracistronic mutation in bacteriophage Q beta RNA. J Mol Biol 1974;89:255-72. Flora SJ. Role of free radicals and antioxidants in health and disease. Cell Mol Biol (Noisy -le-grand) 2007;53:1-2. Fortini P, Pascucci B, Parlanti E, D'Errico M, Simonelli V, Dogliotti E. The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie 2003;85:105371. Gazzoli I, Kolodner RD. Regulation of the human MSH6 gene by the Sp1 transcription factor and alteration of promoter activity and expression by polymorphisms. Mol Cell Biol 2003;23:7992-8007. Gould SJ, Subramani S. Firefly luciferase as a tool in molecular and cell biology. Anal Biochem 1988;175:5-13. Hagmar L, Bonassi S, Stromberg U et al. Chromosomal aberrations in lymphocytes predict human cancer: a report from the European Study Group on Cytogenetic Biomarkers and Health (ESCH). Cancer Res 1998;58:4117-21. 82 Hailer-Morrison MK, Kotler JM, Martin BD, Sugden KD. Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2'-deoxyguanosine lesions in the NF-kappaB promoter element. Biochemistry 2003;42:9761-70. Hasselbach L, Haase S, Fischer D, Kolberg HC, Sturzbecher HW. Characterisation of the promoter region of the human DNA-repair gene Rad51. Eur J Gynaecol Oncol 2005;26:589-98. Hazra TK, Izumi T, Venkataraman R, Kow YW, Dizdaroglu M, Mitra S. Characterization of a novel 8-oxoguanine-DNA glycosylase activity in Escherichia coli and identification of the enzyme as endonuclease VIII. J Biol Chem 2000;275:27762-67. Hazra TK, Kow YW, Hatahet Z et al. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem 2002;277:30417-20. Hazra TK, Izumi T, Kow YW, Mitra S. The discovery of a new family of mammalian enzymes for repair of oxidatively damaged DNA, and its physiological implications. Carcinogenesis 2003;24:155-57. Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair (Amst) 2007;6:470-480. Hecht SS, Hoffmann D. The relevance of tobacco-specific nitrosamines to human cancer. Cancer Surv 1989;8:273-94. Hecht SS. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat Res 1999;424:127-42. Hill CE, Wickliffe JK, Wolfe KJ, Kinslow CJ, Lopez MS, bdel-Rahman SZ. The L84F and the I143V polymorphisms in the O6-methylguanine-DNA-methyltransferase (MGMT) gene increase human sensitivity to the genotoxic effects of the tobacco-specific nitrosamine carcinogen NNK. Pharmacogenet Genomics 2005;15:571-78. 83 Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: a controversial issue. a tribute to Ernst L. Wynder. Chem Res Toxicol 2001;14:767-90. Hoshi S, Hijikata M, Togashi Y et al. Protein C deficiency in a family with thromboembolism and identified gene mutations. Intern Med 2007;46:997-1003. Hsu TC, Johnston DA, Cherry LM et al. Sensitivity to genotoxic effects of bleomycin in humans: possible relationship to environmental carcinogenesis. Int J Cancer 1989;43:403-9. Hu Z, Wang H, Shao M et al. Genetic variants in MGMT and risk of lung cancer in Southeastern Chinese: a haplotype-based analysis. Hum Mutat 2007;28:431-40. Huet A, Parlakian A, Arnaud MC et al. Mechanism of binding of serum response factor to serum response element. FEBS J 2005;272:3105-19. Izumi T, Wiederhold LR, Roy G et al. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology 2003;193:4365. Ji LL. Antioxidant signaling in skeletal muscle: a brief review. Exp Gerontol 2007;42:582-93. Jones JA, Riggs PK, Yang TC et al. Ionizing radiation-induced bioeffects in space and strategies to reduce cellular injury and carcinogenesis. Aviat Space Environ Med 2007;78:A67-A78. Jungst C, Cheng B, Gehrke R et al. Oxidative damage is increased in human liver tissue adjacent to hepatocellular carcinoma. Hepatology 2004;39:1663-72. Karihtala P, Soini Y. Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. APMIS 2007;115:81-103. Kim SH, Bae JS, Holloway JW et al. A polymorphism of MS4A2 (- 109T > C) encoding the beta-chain of the high-affinity immunoglobulin E receptor (FcepsilonR1beta) is 84 associated with a susceptibility to aspirin-intolerant asthma. Clin Exp Allergy 2006;36:877-83. Kiyohara C, Yoshimasu K, Takayama K, Nakanishi Y. EPHX1 polymorphisms and the risk of lung cancer: a HuGE review. Epidemiology 2006;17:89-99. Korolev BG. [Base excision repair of DNA: glycosylases]. Genetika 2005;41:725-35. Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett 2000;476:73-77. Lander ES, Linton LM, Birren B et al. Initial sequencing and analysis of the human genome. Nature 2001;409:860-921. Langen RC, Korn SH, Wouters EF. ROS in the local and systemic pathogenesis of COPD. Free Radic Biol Med 2003;35:226-35. le-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clin Chem 2006;52:601-23. Lefstin JA, Yamamoto KR. Allosteric effects of DNA on transcriptional regulators. Nature 1998;392:885-88. Leong DT, Gupta A, Bai HF et al. Absolute quantification of gene expression in biomaterials research using real-time PCR. Biomaterials 2007;28:203-10. Li C, Hu Z, Lu J et al. Genetic polymorphisms in DNA base-excision repair genes ADPRT, XRCC1, and APE1 and the risk of squamous cell carcinoma of the head and neck. Cancer 2007;110:867-75. Liao CH, Hsiao YM, Hsu CP et al. Transcriptionally mediated inhibition of telomerase of fungal immunomodulatory protein from Ganoderma tsugae in A549 human lung adenocarcinoma cell line. Mol Carcinog 2006;45:220-229. 85 Lu J, Wang LE, Xiong P, Sturgis EM, Spitz MR, Wei Q. 172G>T variant in the 5' untranslated region of DNA repair gene RAD51 reduces risk of squamous cell carcinoma of the head and neck and interacts with a P53 codon 72 variant. Carcinogenesis 2007;28:988-94. Luo LZ, Werner KM, Gollin SM, Saunders WS. Cigarette smoke induces anaphase bridges and genomic imbalances in normal cells. Mutat Res 2004;554:375-85. Mathy-Hartert M, Martin G, Devel P et al. Reactive oxygen species downregulate the expression of pro-inflammatory genes by human chondrocytes. Inflamm Res 2003;52:111-18. Maunders H, Patwardhan S, Phillips J, Clack A, Richter A. Human bronchial epithelial cell transcriptome: gene expression changes following acute exposure to whole cigarette smoke in vitro. Am J Physiol Lung Cell Mol Physiol 2007;292:L1248-L1256. McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annu Rev Biochem 1999;68:255-85. Mohrenweiser H. Survey of polymorphic sequence variation in the immediate 5' region of human DNA repair genes. Mutat Res 2007;616:221-26. Morel Y, Barouki R. Repression of gene expression by oxidative stress. Biochem J 1999;342 Pt 3:481-96. Morland I, Rolseth V, Luna L, Rognes T, Bjoras M, Seeberg E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res 2002;30:4926-36. Mottagui-Tabar S, Faghihi MA, Mizuno Y et al. Identification of functional SNPs in the 5-prime flanking sequences of human genes. BMC Genomics 2005;6:18. Muller K, Gawlik I. Effects of reactive oxygen species on the biosynthesis of 12 (S)hydroxyeicosatetraenoic acid in mouse epidermal homogenate. Free Radic Biol Med 1997;23:321-30. 86 Niess AM, Simon P. Response and adaptation of skeletal muscle to exercise--the role of reactive oxygen species. Front Biosci 2007;12:4826-38. Nishigori C, Hattori Y, Toyokuni S. Role of reactive oxygen species in skin carcinogenesis. Antioxid Redox Signal 2004;6:561-70. Ogbourne S, Antalis TM. Transcriptional control and the role of silencers in transcriptional regulation in eukaryotes. Biochem J 1998;331 ( Pt 1):1-14. Pages V, Fuchs RP. How DNA lesions are turned into mutations within cells? Oncogene 2002;21:8957-66. Pakakasama S, Kanchanakamhaeng K, Kajanachumpol S et al. Genetic polymorphisms of folate metabolic enzymes and toxicities of high dose methotrexate in children with acute lymphoblastic leukemia. Ann Hematol 2007;86:609-11. Paz-Elizur T, Brenner DE, Livneh Z. Interrogating DNA repair in cancer risk assessment. Cancer Epidemiol Biomarkers Prev 2005;14:1585-87. Pereira LH, Pineda MA, Rowe WH et al. The BRCA1 Ashkenazi founder mutations occur on common haplotypes and are not highly correlated with anonymous single nucleotide polymorphisms likely to be used in genome-wide case-control association studies. BMC Genet 2007;8:68. Radisavljevic ZM, Gonzalez-Flecha B. Signaling through Cdk2, importin-alpha and NuMA is required for H2O2-induced mitosis in primary type II pneumocytes. Biochim Biophys Acta 2003;1640:163-70. Sakumi K, Tominaga Y, Furuichi M et al. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res 2003;63:902-5. Schoonbroodt S, Piette J. Oxidative stress interference with the nuclear factor-kappa B activation pathways. Biochem Pharmacol 2000;60:1075-83. 87 Schug J, Overton GC. Modeling transcription factor binding sites with Gibbs Sampling and Minimum Description Length encoding. Proc Int Conf Intell Syst Mol Biol 1997;5:268-71. Shen H, Spitz MR, Qiao Y et al. Smoking, DNA repair capacity and risk of nonsmall cell lung cancer. Int J Cancer 2003;107:84-88. Southam L, Rodriguez-Lopez J, Wilkins JM et al. An SNP in the 5'-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum Mol Genet 2007;16:2226-32. Spielman RS, Bastone LA, Burdick JT, Morley M, Ewens WJ, Cheung VG. Common genetic variants account for differences in gene expression among ethnic groups. Nat Genet 2007;39:226-31. Stepanek J, Vincent M, Turpin PY et al. C-->G base mutations in the CArG box of c-fos serum response element alter its bending flexibility. Consequences for core-SRF recognition. FEBS J 2007;274:2333-48. Strahs D, Barash D, Qian X, Schlick T. Sequence-dependent solution structure and motions of 13 TATA/TBP (TATA-box binding protein) complexes. Biopolymers 2003;69:216-43. Sun Y, Oberley LW. Redox regulation of transcriptional activators. Free Radic Biol Med 1996;21:335-48. Taioli E, Benhamou S, Bouchardy C et al. Myeloperoxidase G-463A polymorphism and lung cancer: a HuGE genetic susceptibility to environmental carcinogens pooled analysis. Genet Med 2007;9:67-73. Takao M, Kanno S, Kobayashi K et al. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J Biol Chem 2002;277:42205-13. Tan W, Miao X, Wang L et al. Significant increase in risk of gastroesophageal cancer is associated with interaction between promoter polymorphisms in thymidylate synthase and serum folate status. Carcinogenesis 2005;26:1430-1435. 88 Tanaka S, Kobayashi I, Utsuki S, Oka H, Yasui Y, Fujii K. Down-regulation of O6methylguanine-DNA methyltransferase gene expression in gliomas by platinum compounds. Oncol Rep 2005;14:1275-80. Thun MJ, Lally CA, Flannery JT, Calle EE, Flanders WD, Heath CW, Jr. Cigarette smoking and changes in the histopathology of lung cancer. J Natl Cancer Inst 1997;89:1580-1586. Van Laere SJ, Van dA, I, Van den Eynden GG et al. NF-kappaB activation in inflammatory breast cancer is associated with oestrogen receptor downregulation, secondary to EGFR and/or ErbB2 overexpression and MAPK hyperactivation. Br J Cancer 2007;97:659-69. van Leeuwen DM, Gottschalk RW, van Herwijnen MH, Moonen EJ, Kleinjans JC, van Delft JH. Differential gene expression in human peripheral blood mononuclear cells induced by cigarette smoke and its constituents. Toxicol Sci 2005;86:200-210. Venter JC, Adams MD, Myers EW et al. The sequence of the human genome. Science 2001;291:1304-51. Verweij CL, Gringhuis SI. Oxidants and tyrosine phosphorylation: role of acute and chronic oxidative stress in T-and B-lymphocyte signaling. Antioxid Redox Signal 2002;4:543-51. Villard J. Transcription regulation and human diseases. Swiss Med Wkly 2004;134:57179. Wang B, Ngoi S, Wang J, Chong SS, Lee CG. The promoter region of the MDR1 gene is largely invariant, but different single nucleotide polymorphism haplotypes affect MDR1 promoter activity differently in different cell lines. Mol Pharmacol 2006;70:267-76. Wang H, Ogawa M, Wood JR et al. Genetic and epigenetic mechanisms combine to control MMP1 expression and its association with preterm premature rupture of membranes. Hum Mol Genet 2008. 89 Wang YP, Tang XJ, Chen XH et al. [Study on cloning of hTERT promoter and its targeting transcriptional activities in telomerase-positive lung cancer cells]. Sichuan Da Xue Xue Bao Yi Xue Ban 2006;37:497-501. Wang ZC, Buraimoh A, Iglehart JD, Richardson AL. Genome-wide analysis for loss of heterozygosity in primary and recurrent phyllodes tumor and fibroadenoma of breast using single nucleotide polymorphism arrays. Breast Cancer Res Treat 2006;97:301-9. Watson JD. The human genome project: past, present, and future. Science 1990;248:4449. Weitberg AB, Corvese D. Oxygen radicals potentiate the genetic toxicity of tobaccospecific nitrosamines. Clin Genet 1993;43:88-91. Wiederhold L, Leppard JB, Kedar P et al. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell 2004;15:209-20. Williams L, Jenkins GJ, Doak SH et al. Fluorescence in situ hybridisation analysis of chromosomal aberrations in gastric tissue: the potential involvement of Helicobacter pylori. Br J Cancer 2005;92:1759-66. Williams TM, Burlein JE, Ogden S, Kricka LJ, Kant JA. Advantages of firefly luciferase as a reporter gene: application to the interleukin-2 gene promoter. Anal Biochem 1989;176:28-32. Wolfe KJ, Wickliffe JK Hill CE Ammenheuser MM Abdel-Rahman SZ. Single nucleotide polymorphisms of the DNA repair gene XPD/ERCC2 alter mRNA expression. Pharmacogenetic Genomics (in press). 2007. Ref Type: In Press Wu X, Zheng YL, Hsu TC. Mutagen-induced chromatid breakage as a marker of cancer risk. Methods Mol Biol 2005;291:59-67. Wu X, Spitz MR, Amos CI et al. Mutagen sensitivity has high heritability: evidence from a twin study. Cancer Res 2006;66:5993-96. 90 Zhang W, Cao YX, He JY, Xu CB. Down-regulation of alpha-adrenoceptor expression by lipid-soluble smoke particles through transcriptional factor nuclear factor-kappaB pathway. Basic Clin Pharmacol Toxicol 2007;101:401-6. Zienolddiny S, Campa D, Lind H et al. Polymorphisms of DNA repair genes and risk of non-small cell lung cancer. Carcinogenesis 2006;27:560-567. 91 VITA Carla Jean Kinslow was born to parents Barbara Jean Newby-Kinslow and Gus Tavis Kinslow in Jeffersonville, Indiana on November 27th, 1969 (just missing Thanksgiving diner). She attended Charlestown High School and subsequently Indiana University Southeast to earn a Bachelor of Arts in Science in 1992. During her sophomore year, she began her research experience by taking a research associate position in the laboratories of Gretchen Kirchner, Ph.D (Doc) and David Taylor, Ph.D. In 1993, she began graduate school at Michigan Technological University as a Challenge Fellow in the laboratory of Gopi Podilla, Ph.D. In 1997, Carla received her Master of Science in Biology and went on to take a position as Scientist at Gene Medicine, Inc. (later, Valentis, Inc.) in The Woodlands, Texas. She then took a research associate position with Dr. Robert Giles in the Vector Core Facility at MD Anderson Cancer Center, Houston, Texas. She then began her own environmental consulting business, Kinslow Consulting, and subsequently created environmental groups for other engineering companies. In 2003, she entered into the Graduate School of Biological Sciences program at the University of Texas Medical Branch, Galveston, Texas. While there, she joined the laboratory of Sherif AbdelRahman, Ph.D. to explore genetic associations to disease risk. While at graduate school, Carla received several honors. In 2003, she received an NIEHS Toxicology Scholarship and subsequently a Toxicology Fellowship in 2005. In 2006 she received the Who’s Who Among American Colleges and Universities award from UTMB. Since 2003, Carla has been invited to present at eight regional and national platform presentations and competed an internship with the Proctor and Gamble Company in Cincinnati, OH. Education B.A. Science, May 1992, Indiana University Southeast, New Albany, IN M.S. Biology, November 1997, Michigan Technological University, Houghton, MI Publications Carla J. Kinslow, Randa A. El-Zein, Courtney E. Hill, Jeffrey K. Wickliffe and Sherif Z. Abdel-Rahman. (2007) Interindividual Variability in NEIL2 Gene Transcription in Human Peripheral Blood Lymphocytes, Carcinogenesis, Submitted Hill CE, Wickliffe JK, Guerin AT, Kinslow CJ, Wolfe KJ, Ammenheuser MM, AbdelRahman SZ. The L84F polymorphism in the O6-Methylguanine-DNA-Methyltransferase (MGMT) gene is associated with increased hypoxanthine phosphoribosyltransferase (HPRT) mutant frequency in lymphocytes of tobacco smokers. Pharmacogenet Genomics. 2007 Sep;17(9):743-53. The L84F and the I143V polymorphisms in the O6-methylguanine-DNAmethyltransferase (MGMT) gene increase human sensitivity to the genotoxic effects of 92 the tobacco-specific nitrosamine carcinogen NNK. Aug;15(8):571-8. Pharmacogenet Genomics. 2005 C.J. Kinslow (1997) Master’s thesis – Molecular Biology of the marine diatom Achnanthesis longipes. Michigan Technological University. G. Kirchner, C.J. Kinslow, G.C. Bloom, and D.W. Taylor (1993). Nonlethal assay system of b-Glucuronidase activity in transgenic tobacco roots. Plant Molecular Biology Reporter 11(4); pages 320-325. 93