Supplementary Materials (Figures) Supplementary Figure 1
advertisement
 Supplementary Figure 1")
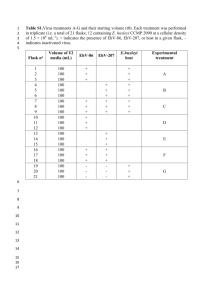
Supplementary Materials (Figures) Supplementary Figure 1. Schematic representation of serial passage model. FV complex was grown in tissue culture in order to produce unpassaged virus stock. This stock was used to begin serial passages. Viruses were collected from the spleens of infected animals and administered to another animal of the same genotype. This was repeated ten times to produce post-passage virus genotypes. During the test phase, each post-passage virus genotype was tested in each of three mouse genotypes; the familiar mouse genotype (i.e. the genotype a virus was serially passaged through) and two unfamiliar genotypes possessing novel MHC and non-MHC polymorphisms. Fitness and virulence estimates among these groups of mice were compared to measure the extent to which viral adaptive responses during serial passage are host-genotypespecific. Supplementary Figure 2. Quantitative PCR (qPCR) Validation. DNA from the spleens of infected animals was extracted using a DNeasy Kit (Qiagen), and the DNA concentration was quantified using a Nanodrop spectrophotometer (Thermo Scientific). Prior to qPCR, all samples were diluted down to approximately 20ng/uL of genomic DNA (gDNA), and 2.5uL of sample was included in each reaction (total 10uL volume). Thus, DNA concentrations per qPCR reaction were standardized to 50ng of total input gDNA. For additional control over variability in input DNA concentrations we also normalized proviral load estimates to GAPD amplicons. Specifically, we used a GAPD primer set, which during amplification from genomic DNA primes from a GAPD pseudogene(s). As we cannot be certain that we are normalizing proviral load estimates to a single copy gene using this approach, we do not report on the number of proviral genomes per host cell, but rather infer the relative abundance of proviral copies per sample. A) To validate our method of standardization we first confirmed that GAPD amplification efficiency curves were similar among the three host genotypes used in this experiment. We detected no statistically significant difference in the efficiency of GAPD amplification among our three host genotypes (MANOVA, adjusted univariate ε, p=0.47). qPCR reactions were run in triplicate (average per dilution shown in figure above)(slopes highlighted in red box). B) We then tested that loading the same concentration of DNA (i.e. ~50ng gDNA) into a reaction yielded consistent crossing point (Cp) values among the three genotypes. As demonstrated above, no significant differences were observed among average GAPD Cp values among the three host genotypes, indicating an invariant number of templates per PCR reaction. (A/J=16.98±0.82, BALB/c=16.29±1.13, and DBA/2J=16.37±1.73)(ANOVA, F 2,27=0.85, 0.44). C) Finally, comparison of PCR product melting peaks (i.e. Tm analysis) also did not show any difference among our three host genotypes (ANOVA, F2,28=0.58, p=0.56 ). This indicates that our GAPD primer set is either priming from the same locus, or priming from different loci that produce similar products across the three host genotypes. Together, these data indicate that our method of standardizing input DNA concentration across samples was appropriate as there is no bias in the GAPD qPCR reaction among our three host genotypes.