EMI_2544_sm_Suppl
advertisement

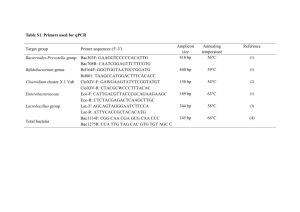
Supplementary Material: Text Diversity analysis methods. Treeclimber (Schloss and Handelsman 2006a) was used to test the hypothesis that the overall structure of two communities is statistically the same. P values indicate the probability that a similar association between phylogeny and sample origin would occur by chance (Schloss and Handelsman 2006a). The mothur software package (Schloss et al. 2009) was used to calculate rarefaction curves, Chao1 (Chao 1984) species diversity of each depth, as well as cross-comparisons of community structure using abundance-based Jaccard (Yue et al. 2001) and Theta (Yue and Clayton 2005) diversity indices. Both individual community Chao1 estimates, and estimates of the Chao1 diversity shared between two communities (shared Chao1) were calculated. The abundance-based Jaccard index measures the intersection of the combined assemblage of phylotypes, and can be considered the probability that given a phylotype is present in one assemblage, it is present in both assemblages (Schloss and Handelsman 2006b). By contrast, ThetaYC corrects for the differences in relative abundance of phylotypes in each assemblage, taking into account both the incidence of shared phylotypes and the similarity of their relative abundances in each assemblage. Thus, it can be considered a percentile measure of the similarity of community structures. Archaeal and Bacterial species richness. The Archaeal and Bacterial clone libraries differed widely in the species diversity as estimated by rarefaction analysis (Figure S2) and Chao1 indices (Table S3A,B). For bacterial community structure comparisons, operational taxonomic units (OTU) were based on 3% (Stackebrandt and Goebel 1994) rather than 1% sequence divergence used for the Archaea (Stackebrandt and Ebers 2009), due to the high sequence richness of the bacterial dataset. Archaeal diversity appeared to be fully sampled by four to ten 1% OTUs for all depths. In contrast, rarefaction curves of Bacteria did not reach saturation at any sediment depth (Figure S2). Chao1 estimates showed a similar trend and indicated 3 to 10 archaeal species at 1% OTU definition, but several tens to several hundreds of bacterial 3% OTU definition (Table S3B). There are three possible explanations for this conspicuous difference in bacterial and archaeal diversity: 1) If these results are a reasonable approximation of the true diversity of the Archaea and Bacteria in situ, highly restricted archaeal diversity might indicate that the oligotrophic sediment biome represents an extreme habitat for Archaea, where only a few specialists thrive. This is consistent with the observation that the SPG12 archaea are distinct from the archaeal lineages abundant at organic-rich margin sites (e.g., Biddle et al. 2006, 2008; Inagaki et al. 2006; Fry et al. 2008); in these habitats, they constitute a major or even the dominant portion of the overall microbial biomass (Lipp et al. 2008). 2) If Archaea occur in significantly lower abundance than Bacteria at SPG12, their low DNA and target gene concentration leads to stochastically biased PCR amplifications, diminishing the species diversity detectable in clone libraries. Yet given a conservative 25% recovery of cellular DNA from ~10 mL of sediment containing 350,000 to 10,000,000 cells per mL of sediment, and using 2% of this recovered DNA in each PCR reaction, at least 21,000 bacterial and archaeal 16S rRNA gene molecules should be present in each PCR (Table S4). Assuming the 2/5 ratio of Crenarchaeota to Bacteria found in the abyssal North Pacific water column (Karner et al. 2001), this translates into a minimum archaeal 16S gene number of ca. 4000 per PCR. To minimize the potential effect of PCR bias on clone library diversity, three PCR reactions were combined for each cloning procedure, PCR cycle number was kept as low as possible, nested PCR was avoided, and multiple precautions were taken to maximize DNA extraction yields (see Methods). The potential number of genomes per PCR reaction, based on sample cell counts and the assumption of 25% DNA extraction efficiency, did not suggest a direct relationship between PCR DNA concentrations and clone library composition and diversity (Table S4). The impact of sediment-derived PCR inhibitors on clone library diversity is yet unknown. 3) A third possibility, not mutually exclusive of the previous two, is that the archaeal 16S rRNA genes recovered from SPG12 may have substitutions in key primer sites, with only rare phylotypes being amenable to PCR using 8f/1492r primers. Highly selective PCR amplification of a few matching archaeal phylotypes from otherwise mismatched groups would produce nearly identical archaeal sequences multiple times from different depths, possibly under widely varying geochemical regimes (Figure 1; Table S2). For example, the DSEG-2 phylotype SPG12_401_411_A1, comprising ~38% of clones from 401-411 cmbsf, also made up 5% of clones at 210-220 cmbsf; and the MBG-A2 phylotypes were found in abundance at both 273-283 and 343-353 cmbsf. The frequent recovery of identical archaeal phylotypes from different samples and PCR reactions is hard to explain by stochastic PCR bias only, but would be congruent with primer bias. Mismatched PCR primers might be one of the reasons why representatives of SPG12 archaeal lineages are rare in public databases (Table S1). More generally, the effects of primer bias can be seen in the divergent outcomes of mutually independent archaeal 16S rRNA gene PCR analyses of the same subsurface sediments (Teske and Sørensen 2008). Yet, the 16S rRNA gene primer pair used in this study had been adjusted for greater archaeal coverage by shortening the forward primer sequence from E.coli positions 8 to 27, to positions 8 to 24 (Teske et al. 2002), to avoid mismatches in these critical nucleotides at the 3’-end of this frequently targeted priming region that were repeatedly found in database searches especially among euryarcheota (A. Teske, unpublished results). In two field tests, the modified archaeal primer pair recovered a new euryarchaeotal lineage (Teske et al. 2002), subsequently named the Guaymas Euryarchaeotal Group (Dhillon et al. 2005). Ultimately, the explanation for the low Archaeal diversity may in fact be a combination of all three alternatives described above. Newly developed, multiple PCR primer sets, as well as PCR-independent methods such as whole-genome amplification (Biddle et al. 2008), are needed to characterize the unique microbial communities in oligotrophic marine deep sediments. At this stage, the SPG12 results showing low archaeal species diversity, and the resulting archaeal comparative statistics should be interpreted with caution. References for supplementary text: Biddle, J.F., Lipp, J.S., Lever, M.A., Lloyd, K.G., Sørensen, K.B., Anderson, R., Fredricks, H.F., Elvert, M., Kelly, T.J., Schrag, D.P., Sogin, M.L., Brenchley, J.E., Teske, A., House, C.H., and Hinrichs. K.-U. (2006) Heterotrophic Archaea dominate sedimentary subsurface ecosystems off Peru. Proc Natl Acad Sci USA 103: 3846-3851. Biddle, J.F., Fitz-Gibbon, S., Schuster, S.C., Brenchley, J.E., and House, C.H. (2008) Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc Natl Acad Sci USA 105: 10583-10588. Chao, A. 1984. Non-parametric estimation of the number of classes in a population. Scandinavian Journal of Statistics 11:265–270. Dhillon, A., Lever, M.A., Lloyd, K.G., Albert, D.B., Sogin, M.L., and Teske, A. (2005) Methanogen Diversity Evidenced by Molecular Characterization of Methyl Coenzyme M Reductase A (mcrA) Genes (mcrA) in Hydrothermal Sediments of the Guaymas Basin. Appl Environ Microbiol 71:4592-4601. Fry, J.C., Parkes, R.J., Cragg, B.A., Weightman, A.J., and Webster, G. (2008) Prokaryotic biodiversity and activity in the deep subseafloor biosphere. FEMS Microbiol Ecol 66: 181-196. Inagaki, F., Nunoura, T., Nakagawa, S., et al. (2006) Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc Natl Acad Sci USA 103: 2815-2820. Karner, M.B., DeLong, E.F., and Karl, D.M. (2001) Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409: 507-510. Lipp, J.S., Morono, Y., Inagaki, F., and Hinrichs, K.-U. (2008) Significant contribution of Archaea to extant biomass in marine subsurface sediments. Nature 454: 991-994. Schloss, P.D., and Handelsman, J. (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71: 1501-1506. Schloss, P.D., and Handelsman, J. (2006a) Introducing TreeClimber, a test to compare microbial community structures. Appl Environ Microbiol 72: 2379-2384. Schloss, P.D., and Handelsman, J. (2006b) Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72: 6773-6779. Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., Lesniewski, R.A., Oakley, B.B., Parks, D.H., Robinson, C.J., Sahl, J.W., Stres, B., Thallinger, G.G., van Horn, D.J., Weber, C.F. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537-7541. Stackebrandt, E., and Ebers, J. (2009) Taxonomic parameters revisited: tarnished gold standards. Microbiology Today, November 6 issue,152-155. Stackebrandt, E., and Goebel, B.M. (1994) Taxonomic Note: A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44:846-849. Teske, A., Hinrichs, K.-U., Edgcomb, V., De Vera Gomez, A., Kysela, D, Sylva S. P., Sogin, M.L., and Jannasch, H.W. (2002) Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl Environ Microbiol 68: 1994-2007. Teske, A., and Sørensen, K.B. (2008) Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J 2: 3-18 Yue, J.C., Clayton, M.K., and Lin, F.C (2001) A nonparametric estimator of species overlap. Biometrics 57: 743-749. Yue, J.C., and Clayton, M.K. (2005) A similarity measure based on species proportions. Commun Stat Theory Meth 34: 2123-2131. Supplementary material: Figures Figure S1. Neighbor-Joining phylogeny of all full-length 3% OTU representatives for SPG12 Planktomycetes (A), Chloroflexi (B), and other bacterial phyla (C, D, E), based on ~1200-bp alignments of 16S rRNA sequences. A maximum-composite likelihood substitution model with gamma-corrected site rates was used in MEGA4. Support for clades was estimated by 1000 interior branch test replicates. 3% representative phylotypes defined on partial reads for the 153-163 depth layer were not included in phylogenies (Table S2). References for previously defined uncultured clades are as follows: Fig. S1(A): CPla-3, Köhler et al. (2008). Fig S1(B): SAR202, Giovannoni et al. (1996); SAR202 subclusters, Morris et al. (2004); Chloroflexi clades I-IV, Inagaki et al. (2006). S1(C): NKB19, Li et al. (1999), Hugenholtz et al. (2002); SAR324, Wright et al. (1997); Termite Group I, Ohkuma and Kudo (1996); OP8, Hugenholtz et al. (1998); NTB2, Reed et al. (2002). Fig. S1(E): SAR11, Giovannoni et al. (1990); SARmet-5, Teske et al. (2011); JTB255, Li et al. (2009). Fig. S1(D): SAR406, Gordon and Giovannoni (1996); WS3, Dojka et al. (1998); NT-B9, Reed et al. (2002); BRC1 (Derakshani et al. 2001). Figure S2. Rarefaction analysis at 1% OTU resolution for Archaea (gray symbols) and Bacteria (black symbols). Table S1. Representative phylotypes for archaea in the SPG12 sediment column, based on 1% OTU definition. The accession number, the best BLAST hit, the phylogenetic classification, and the number of clones in different sediment horizons are listed for each phylotype. Table S2. Representative phylotypes for bacteria in the SPG 12 sediment column, based on 3% OTU definition. The accession number, phylogenetic classification, and the number of clones in different sediment horizons are listed for each phylotype. Table S3. Archaeal and Bacterial diversity indices for SPG12 clone libraries. Furthestneighbor diversity analyses for SPG12 Archaea (A) and Bacteria (B) were calculated in mothur (Schloss et al. 2009), using the furthest-neighbor approach and an OTU definition of 3%. For comparative indices, the first row shows the shared Chao1 estimated diversity (Chao 1984), the second row lists abundance-based Jaccard (Yue et al. 2001), and the third ThetaYC similarity (Yue and Clayton 2005). Also listed for Bacteria is the Shannon diversity index and the Pielou evenness index, both calculated using phylum as the relevant taxonomic unit. . Table S4. Relative comparison of potential DNA concentrations in PCR reactions, based on sediment cell densities, volume of sediment extracted, and volume of PCR extract used per PCR reaction. 3% OTU bacterial and 1% OTU archaeal Chao1 diversity is provided for comparison. 25% DNA extraction efficiency is assumed for the purposes of this comparison. Figure S1 References: Chao, A. (1984) Non-parametric estimation of the number of classes in a population. Scandinavian Journal of Statistics 11:265–270. Derakshani, M., Lukow, T., and Liesack, W. (2001) Novel bacterial lineages at the (Sub)Division level as detected by signature nucleotide-targeted recovery of 16S rRNA genes from bulk soil and rice roots of flooded rice microcosms. Appl Environ Microbiol 67: 621-631 Dojka, M.A., Hugenholtz, P., Haack, S.K., and Pace, N.R. (1998) Microbial diversity in a hydrocarbon- and chlorinated-solvent contaminated aquifer undergoing intrinsic bioremediation. Appl Environ Microbiol 64: 3869-3877. Giovannoni, J.A., Britschgi, T.B., Moyer, C.L., and Field, K.G. (1990) Genetic diversity in Sargasso Sea bacterioplankton. Nature 345:60-63. Giovannoni, J.A., Rappé, M.S., Vergin, K.L., and Adar, N.L. (1996) 16S rRNA genes reveal stratified open ocean bacterioplankton populations related to the green non-sulfur bacteria. Proc Natl Acad Sci USA 93: 7979-7984. Gordon, D.A and Giovannoni, S.J. (1996) Detection of stratified microbial populations related to Chlorobium and Fibrobacter species in the Atlantic and Pacific Oceans. Appl. Environ. Microbiol. 62:1171-1177. Hugenholtz, P., Pitulle, C., Hershberger, K.L., Pace, N.R. (1998) Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol 180: 366–376. Hugenholtz, P. (2002) Exploring prokaryotic diversity in the genomic era. Genome Biol 3: reviews0003.1-reviews0003.8. Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M.A., Lauer, A., Suzuki, M., Takai, K., Delwiche, M., Colwell, F.S., Nealson, K.H., Horikoshi, K., D'Hondt, S.L., and Jørgensen , B.B. (2006) Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc Natl Acad Sci USA 103:2815-2820. Köhler, T., Stingl, U., Meuser, K., and Brune, A. (2008) Novel lineages of Planctomycetes densely colonize the alkaline gut of soil-feeding termites (Cubitermes spp.). Environ Microbiol 10: 1260-1270. Li, L., Kato, C., and Horikoshi, K. (1999) Bacterial diversity in deep-sea sediments from different depths. Biodiv Conserv 8: 659-677. Li, H., Yu, Y., Luo, W., Zeng, Y., and Chen, B. (2009) Bacterial diversity in surface sediments from the Pacific Arctic Ocean. Extremophiles 13: 233-246. Morris, R.M., Rappé, M.S., Urbach, E., Connon, S.A., Giovannoni, S.J. (2004) Prevalence of the Chloroflexi-related SAR202 bacterioplankton cluster throughout the mesopelagic zone and deep ocean. Appl Env Microbiol 70: 2836-2842. Ohkuma, M., and Kudo, T. (1996) Phylogenetic diversity of the intestinal bacterial community in the termite Reticulitermes speratus. Appl Environ Microbiol 62: 461-468. Reed, D.W., Fujita, Y., Delwiche, M.E., Blackwelder, D.B., Sheridan, P.P., Uchida, T., and Colwell, F.S. (2002) Microbial communities from methane hydrate-bearing deep marine sediments in a forearc basin. Appl Environ Microbiol 68:3759-3770. Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., Lesniewski, R.A., Oakley, B.B., Parks, D.H., Robinson, C.J., Sahl, J.W., Stres, B., Thallinger, G.G., van Horn, D.J., and Weber, C.F. (2009) Introducing mothur: opensource, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537-7541. Teske, A., Durbin, A.M., Ziervogel, K., Cox, C., and Arnosti, C. (2011) Microbial community composition and function in permanently cold seawater and sediments from an Arctic fjord of Svalbard. Appl Environ Microbiol 77:2008-2018. Wright, T.D., Vergin, K.L., Boyd, P.W., and Giovannoni, S.J. (1997) A novel delta- subdivision proteobacterial lineage from the lower ocean surface layer. Appl Environ Microbiol 63: 1441-1448. Yue, J.C., Clayton, M.K., and Lin, F.C (2001) A nonparametric estimator of species overlap. Biometrics 57: 743-749. Yue, J.C., and Clayton, M.K. (2005) A similarity measure based on species proportions. Commun Stat Theory Meth 34: 2123-2131.