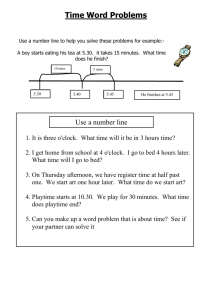
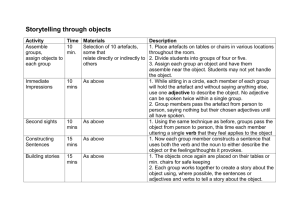
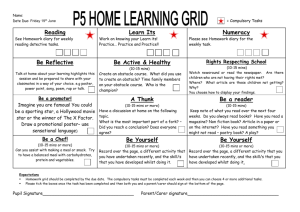
Word file (27 KB )
advertisement
")
1 Supplementary material Individual mosquitoes were homogenised in 1001 of ice-cold phosphate buffer (50mM, pH 7.4), which had previously been treated with diethyl pyrocarbonate (DEPC) (Sigma) in order to inhibit RNase activity. The homogenate was centrifuged at 12,000g, 4oC for 5 mins and the supernatant transferred to a clean Eppendorf centrifuge tube. Esterase activity was determined by adding 200l of 1mM para-nitrophenyl acetate to 10l of insect supernatant. Activity was measured continuously for 2 mins at 405nm in a Molecular Devices thermomax plate reader. Assays were carried out in triplicate for each mosquito. As the digesting bloodmeal affected mosquito protein analysis, protein concentrations were determined in 10l of supernatant from 20 non-blood fed females by the method of Bradford using the BioRad protein assay kit. Mean values were then used to convert esterase values into specific activities. After esterase analysis, total RNA was extracted from the remaining mosquito supernatant. Each homogenate was mixed with 150l of TRI reagent (Sigma) and the mixture incubated at room temperature for 5 mins. Residual insoluble insect debris was removed by centrifugation (12,000g, 4oC for 10 mins) and the supernatant transferred to a fresh centrifuge tube. The supernatant was mixed vigorously with 30l of chloroform and incubated at room temperature for 5 mins before being centrifuged as above. The aqueous layer was placed in a clean tube and the RNA precipitated using 70l of isopropanol. RNA was pelleted by centrifugation at 12,000g, 4oC for 15 mins and washed twice with 75% ethanol. RNA pellets were air dried, resuspended in 20ml of DEPC treated distilled water and treated with DNase (Promega) to ensure the RNA was DNA free. All reagents for cDNA synthesis were obtained from Gibco BRL unless otherwise stated. 10l of RNA was incubated with 0.5 g oligo (dT)15 (Promega) at 70oC for 10 mins. Samples were placed on ice for 2 mins and the cDNA synthesis mixture (1 x first strand buffer, 7.14mM DTT, 0.36mM dNTPs) was added. Reaction mixtures were incubated for 2 mins at 42oC, 200 Units of superscript RT were added and incubation continued for a further 50 mins. The reaction was terminated by heating at 70oC for 15 mins. 2 [Supplementary material, Cont/d….] Quantitative PCR was carried out on a Roche Light Cycler. PCR reaction mixtures comprised 1l template cDNA, 0.1mM each primer, 1 x reaction buffer (containing SYBR Green, Taq DNA polymerase, dNTP mix, 10mM MgCl2 [Roche]). PCR primers were obtained from Sigma Genosys. A standard curve was constructed using the Wuchereria bancrofti primers NV1 5’ CGTGATGGCATCAAAGTAGCG 3’ and NV2 5’ CCTCAACTTACCATAAGACAAC 3’ (Nicolas & Plichart 1997) and serial dilutions of parasite DNA kindly provided by Dr P. Esterre, French Polynesia). Quantitative PCR used 40 cycles of 95oC 1 second, 55o C 2 seconds, 72oC 10 seconds. Data were acquired at the end of each cycle by measuring fluorescence at 80o C. Sample cDNA reactions were run using the same conditions and parasite cDNA was quantified using the standard curve. Results were standardised using an internal actin control for each sample. Actin primers ActF 5’ GCGACGAGGCCCAGAGCA 3’ and ActR 5’ TCCAGCGCGACGTAGCACAG 3’ were designed from Salazar et al (1994). Parameters for the actin QPCR were 40 cycles of 95oC 1 second, 62oC for 5 seconds, 72 oC for 10 seconds. Data were acquired at the end of each cycle by measuring fluorescence at 72 oC. Infective L3 larvae were detected in 12 day old mosquitoes by microscopy after feeding on W. bancrofti infected blood with an intermediate level of parasitaemia. Feeding resulted in < 1% mortality of adult female mosquitoes, which was comparable with controls. Acknowledgements This work was funded by a one year ROPA award from The Medical Research Council UK and a small grant from The Royal Society to JH and MP. 3