View
advertisement

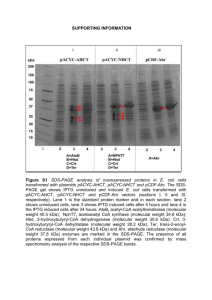
Supplementary Material Chemicals and reagents L-asparagine (L-Asn), flavine adenine dinucleotide (FAD), nicotinamide adenine dinucleotide (NADH), horseradish peroxidase (HRP type X, 386 U/mg, Mr ~ 44 kDa), glutamate dehydrogenase from bovine liver (GDH, 44 U/mg, Mr ~ 56 kDa), αketoglutarate, o-dianisidine, L-aspartate (L-Asp), and human serum (from male AB plasma) were obtained from Sigma Aldrich (St. Louis MO, USA). Amplex Red (AR) was purchased from Cayman chemicals (Michigan, USA). Yeast extract, peptone, agar, ampicillin, and Isopropyl-beta-D-thiogalactoside (IPTG) were purchased from Applichem (Gatersleben, Germany). KAPA HiFi polymerase was obtained from PeqLab (Erlangen, Germany), restriction enzymes and T4 DNA ligase from NEB (Ipswich, MA, USA). Gel extraction and PCR purification kits, and nickel agarose for affinity purification of polyhistidine-tagged proteins were from Macherey Nagel (Düren, Germany), plasmid purification kit from Fermentas (Thermo Fisher Scientific, Germany), genomic DNA preparation kit from Qiagen (Hilden, Germany), gel filtration chromatography column Superdex 200 from Pharmacia/GE Health Care Life Sciences (Uppsala Sweden). Coomassie Brilliant Blue G-250 (Bradford reagent) for protein assays was from Roth (Karlsruhe, Germany). Cloning, expression and purification of L-aspartate oxidase and periplasmic Lasparaginase from E.coli. Since L-aspartate oxidase (L-AspOx) is commercially not available, we cloned the E.coli gene (nadB) and produced milligram amounts of the enzyme recombinantly. The open reading frame (ORF) of L-AspOx consisting of 1623 bp, was amplified by PCR using as template genomic DNA isolated from the E.coli XL1-Blue strain. Unique NdeI and BamHI restrictions sites (underlined below) were incorporated at the 5’- and 3’- ends of the respective oligonucleotides (5’-GGAATTCCATATGAATACTCTCCCTGAACATTC3’, and 5’-CGCGGATCCTTATCTGTTTATGTAATGATTGC-3’). The PCR mixture (50 μL) consisted of 1 μg genomic DNA, oligonucleotide mix (10 pmol each), KAPA high fidelity buffer, dNTPs (0.2 mM each), and 1 Unit KAPA HiFi DNA polymerase. The reaction was performed at initial temperature of 95 °C for 3 min, followed by 25 cycles of the next steps: at 98 °C for 20 s, 60 °C for 60 s, and 72 °C for 30 s. The reaction was terminated after a 3’-end polishing step of 5 min at 72 °C. The PCR product was agarose gel-purified, digested with NdeI and BamHI HF, and ligated overnight at 16 °C into a pET14b-SUMO vector [1] using T4 DNA ligase. The ligation mixture was used to transform DH5α E.coli cells, and positive clones were first identified by colony PCR screening. Extracted plasmid DNA was digested with NdeI and BamHI HF to verify correct size of the cloned fragment, and finally the gene insert was sequenced (SeqLab, Goettingen, Germany). The expressed protein construct includes an N-terminal six-histidine tag, followed by a yeast SUMO (Small Ubiquitin-related MOdifier) tag which has proven to ameliorate heterologous protein solubility and stability [2]. E.coli BL21(DE3) C41 cells (Lucigen, Madison, USA) were cultured overnight at 37 °C in LB medium supplemented with 200 μg/ml ampicillin. A fraction of this culture was used to inoculate fresh 2xYT culture medium (dilution 1:100) supplemented with 200 μg/ml ampicillin. When O.D. 600 reached ~ 0.5-0.7, expression was induced by adding IPTG to a final concentration of 1 mM. After incubation at 37 °C for 8 hours, the cells were harvested by centrifugation (20 min at 5,000g), resuspended in nickel agarose binding buffer A (50 mM Na 2HPO4, 0.5 M NaCl, 10 mM imidazole, pH 8), and ultimately lysed by ultrasonication. The cell lysate was centrifuged (45 min at 17,200g), the resulting supernatant mixed with preequilibrated nickel agarose beads, and incubated at 4 °C for 3 hours in a rotating beaker. The slurry mixture was filled in 5 mL polypropylene columns, dried by gravity, and the agarose beads were then washed with 25 bed volumes of buffer B (50 mM Na2HPO4, 1 M NaCl, 25 mM imidazole, pH 8). Bound protein was eluted from the column by applying 300 mM imidazole in buffer B. All purification steps were performed at 4 °C in a cold room. Eluted fractions were mixed, and buffer was exchanged against 50 mM Na2HPO4, 100 mM NaCl, pH 7.5, using a PD-10 column (GE Healthcare). Eluted protein was incubated with yeast SUMO protease (molar ratio 1:100) at 30 °C for 2 hours to cleave the N-terminal tag. This treatment resulted in the final intact L-AspOx with predicted Mr ~ 60.3 kDa (apparent Mr on SDS-PAGE ~ 60 kDa). The protein solution was concentrated using ultrafiltration tubes, 10,000 MWCO (Sartorius, Goettingen, Germany). In the final purification step, the enzyme was passed through a Superdex 200 column to remove the cleaved tag, SUMO protease, and impurities remaining after the affinity purification step. Protein purity, as evaluated by SDS-PAGE, was more than 95%. The protein was aliquoted, mixed with glycerol (25% final concentration), and stored at -20 °C until further use. The overall protein yield for LAspOx was estimated to be approximately 40 mg per L of 2xYT growth medium with a specific activity ~ 0.05 U/mg as determined by applying the o-dianisidine assay described elsewhere [3], which was also used for the kinetic characterization of this enzyme (Fig. S3). Periplasmic mature E.coli L-ASNase (ansB) was cloned using the primer pair 5’GGAATTCCATATGTTACCCAATATCACCATTTTAGC-3’ and 5’- CGCGGATCCTTAGTACTGATTGAAGATCTGCTGG-3’, expressed, and purified in a similar way. The protein amount obtained out of 1 L 2xYT medium was ~ 15 mg with a specific activity of 1 U/mg as determined by the NADH-dependent assay. The predicted Mr of the mature E.coli L-ASNase is 34.5 kDa, while on SDS-PAGE it appears to run somewhat faster (~ 32 kDa). Figures Figure S1. SDS-PAGE (15%) analysis of purified E.coli L-AspOx (calculated Mr: 60.3 kDa). Figure S2. SDS-PAGE (15%) analysis of purified mature E.coli L-ASNase (calculated Mr: 34.5 kDa). Figure S3. Steady-state turnover rates (kobs) of L-AspOx determined by applying the chromogenic o-dianisidine-based assay [3]. Reactions were performed at 25 °C in 1 ml PBS, containing 20 μM FAD, 100 nM HRP, 10 μg/ml o-dianisidine, 65 nM (~ 5 μg) LAspOx, and variable amounts of substrate. Kinetic constants (see text) were calculated by non-linear regression of the Michaelis-Menten (Igor Pro, Wavemetrics). Measurements were performed in triplicate (n = 3), and values are expressed as means ± SD. Figure S4. Fluorescence intensity as a function of AR concentration. The L-Asn concentration was kept constant at 1 μM, while L-ASNase concentration was ~ 4.6 nM. The fluorescence signal was recorded after 30 min in each case, (n=3, ± SD) Figure S5. Time course of the L-ASNase reaction monitored at discrete time points using the fluorescence assay. Concentrations: 10 μM L-Asn, 50 μM AR, ~ 4.6 nM LASNase, (n=3, ± SD). Error bars are not shown when they are smaller than the symbols. Figure S6. Real-time monitoring of the three-step enzymatic reaction to detect Lasparaginase activity using a plate-reader (Molecular Devices, SpectraMax Paradigm; wavelength settings were 532 nm for Ex, and 592 nm for Em). The four reactions took place simultaneously in a 96-well plate in a final volume of 100 μL at the following concentrations: 10 μM L-Asn, 50 μM AR; L-ASNase concentrations were as follows: (●): 0 nM, (■): 1 nM, (▲): 2 nM, and (◇): 4 nM. Figure S7. Enzyme-concentration response using human serum supplemented with LASNase. 200 μL of human serum were spiked with 50 μg L-ASNase previously diluted in PBS, and mixed thoroughly. Subsequently, aliquots were used to detect enzymatic activity in a final volume of 100 μL using standardized conditions as described in the main text for the fluorescence mode, except from the following modifications of concentrations and settings: 100 μM L-Asn and 500 μM AR, Ex. bandwidth at 1 nm, while Em. bandwidth was kept at 2.5 nm. Fluorescence signals were corrected by subtracting the control values (no L-Asn) from all samples, (n=3, ± SD). Figure S8. Steady-state kinetics of L-ASNase applying the NADH-dependent assay. Reactions were performed at 25 °C in 1 ml PBS, containing 0.25 mM NADH, 0.25 mM α-ketoglutarate, 35 nM GDH and 7 nM (~ 250 ng/ml) L-ASNase. Data points are represented as means of triplicate measurements, and error bars are standard deviations of three separate reactions. Kinetic constants were calculated by non-linear regression of the Michaelis-Menten equation (Igor Pro, Wavemetrics) and are shown in Table S1. Table S1. Steady state-kinetic parameters (n=3, ± S.D.) for E.coli L-ASNase determined by applying the AR or the NADH-dependent assay. Kinetic parameter Amplex Red assay NADH assay Km 22 ± 1.2 (μM) 21 ± 1.5 (μM) kcat 12.1 ± 0.5 s-1 11.5 ± 0.4 s-1 kcat/Km 5.5 x 105 M-1 s-1 5.4 x 105 M-1 s-1 Z’ factor determination In order to evaluate and validate the newly developed assay for L-ASNase, we determined the Z’ factor [4] which is defined as follows: where μc+ and μc- are the means of the positive and negative control values, respectively, and σc+ and σc- are the standard deviations of the corresponding signals. The statistical parameters were calculated from six samples containing 10 μM Lasparagine for the positive control, and six ones which contained all components except from L-Asn for the negative control. The final reaction solution contained 3.5 μΜ L- AspOx, 10 μΜ FAD, 100 nM HRP, 50 μΜ AR and 4.6 nM L-ASNase (~ 16 ng/0.1 ml). The Z’ factor was determined to be 0.77 suggesting that this is a very sensitive and reproducible assay. Some Notes and Tips 1) L-AspOx, a monomeric enzyme, has been highly overexpressed recombinantly in E.coli. Instead of IPTG, one could use equally efficiently the cheaper “auto-induction” medium which contains lactose as inducer. We found similar expression yields (>40 mg/L 2XYT medium) in both cases, using the pET-14b-SUMO vector. Such yields could be easily achieved not only when BL21(DE3) C41 was used as expression host, but also with other conventional E.coli strains such as BL21(DE3), BL21(DE3) pLysS, or BL21(DE3) Rosetta. 2) AR is a light-sensitive dye, and therefore it is essential to avoid its exposure to light. We observed that the reaction mixture (without L-ASNase to trigger the reaction) remains stable, exhibiting a very low background fluorescence signal, for 3 hours at room temperature in dark Eppendorf tube. 3) FAD is a crucial co-factor for the activity of L-AspOx; it is non-covalently bound to the enzyme with 1:1 stoichiometry. However, it is not tightly bound (K ~ 1 μM), and therefore a 3-fold molar excess of FAD provides a more stable environment for the activity of L-AspOx. 4) Fresh preparation of all buffers is advantageous, as this can diminish undesired background activity. A noteworthy example is the L-Asn stock solution. L-Asn, when stored for long periods even at 4 °C, can spontaneously hydrolyze to L-Asp and ammonia. This would cause a considerably higher initial background activity, since the presence of L-Asp would initiate reaction steps two and three of the coupledenzyme system. Furthermore, in a series of experiments, we observed that AR remained stable for 4 months when dissolved in DMSO at a final concentration of 50 mM. 5) For the purpose of detection of L-ASNase activity in serum, it is preferable to use saturated L-Asn concentrations (for E.coli L-ASNase > 80 μM) and consequently higher AR concentrations (five fold higher than L-Asn). This will decrease the background reaction and improve the signal-to-noise ratio. References [1] J. Nomme, Y. Su, M. Konrad, A. Lavie, Structures of Apo and Product-Bound Human L-Asparaginase: Insights into the Mechanism of Autoproteolysis and Substrate Hydrolysis, Biochemistry 51 (2012) 6816−6826. [2] T. Panavas, C. Sanders, T. R. Butt, SUMO Fusion Technology for Enhanced Protein Production in Prokaryotic and Eukaryotic Expression Systems, Methods in Molecular Biology, SUMO Protocols 497 (2009) 303-317. [3] G. Tedeschi, S. Ronchi, T. Simonic, C. Treu, A. Mattevi, A. Negri, Probing the active site of L-Aspartate oxidase by site-directed mutagenesis: Role of basic residues in fumarate reduction, Biochemistry 40 (2001) 4738-4744. [4] J.H. Zhang, T.D.Y. Chung, K.R. Oldenburg, A simple statistical parameter for use in evaluation and validation of high throughput screening assays, J. Biomol. Screen. 67 (1999) 67-73.