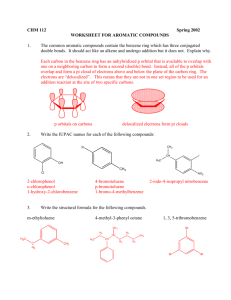
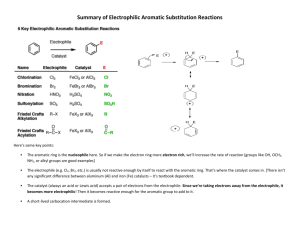
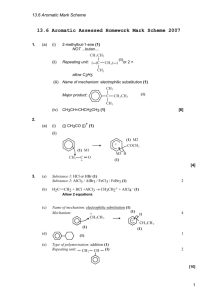
Chemistry of Aromatic Compounds: Course Module
advertisement

KENYATTA UNIVERSITY INSTITUTE OF OPEN LEARNING SCH 302: CHEMISTRY OF AROMATIC COMPOUNDS DR. A.W. WANYONYI DR. A.K. MACHOCHO CHEMISTRY DEPARTMENT CHEMISTRY OF AROMATIC COMPOUNDS PREFACE/GENERAL INTRODUCTION TO SCH 302 The course deals with benzene, its derivatives and other benzenoid compounds. Structure of benzene, aromaticity of benzene and related compounds will form the first part of the course. Nomenclature of benzene and its derivatives are dealt with in details since this will form some of the products of the reactions in the lessons that follow. Mechanistic approach is employed to explain chemical reactions of benzene and its derivatives. Predominant electrophilic aromatic substitution reactions patterns, effect of substituent on the rates of reaction and orientation of incoming substituent are discussed. Reactions of benzenoids are discussed in the light of similarity in chemical behavior to benzene. It will be noted that many compounds will be synthesized in the course of discussing these reactions as the end products Key words in the relevant lessons and problems to enhance understanding or as set induction to lesson to be covered next and give practice to students are provided in the boxes. Each chapter has practice questions at the end and solutions are provided as an illustration on how to approach similar problems. Practicals in the appendix are meant to illustrate specific reactions in the actual conditions applied in the preparation of aromatic compounds especially those covered in the module. CONTENTS Chapter 1: Structure of benzene and aromaticity 1.0 Introduction 1.1 Historical background on benzene 1.2 Structure of benzene 1.3 Characteristics of benzene ring 1.4 Aromaticity and non-aromaticity 1.5 Benzenoid aromatic and non-benzenoid compounds 1.6 Some of aromatic compounds in natural systems 1.7 Summary 1.8 Questions and solutions Chapter 2: Nomenclature of substituted benzenes and other benzenoid compounds 2.0 Introduction 2.1 Nomenclature of monosubstituted benzene 2.2 Nomenclature of substituted benzene 2.3 Nomenclature of other benzene derivatives 2.4 Nomenclature of substituted naphthalenes 2.5 Nomenclature of anthracenes 2.6 Nomenclature of phenanthrenes 2.7 Summary 2.8 Questions and solutions Chapter 3: Reactions of benzene 3.0 Introduction 3.1 Reduction of benzene 3.2 Electrophilic aromatic substitution (EAS) of benzene: A general mechanism 3.3 Halogenation 3.4 Nitration 3.5 Sulphonation 3.6 Friedel-Craft alkylation 3.7 Friedel–Craft acylation 3.8 Nitrosation 3.9 Summary 3.10 Questions and solutions Chapter 4: Reactions of benzene derivatives 4.0 Introduction 4.1 Effect of substituents on EAS patterns/orientation 4.2 Groups that donate electrons to the ring 4.3 Effect of groups that donate by induction and release electrons by resonance 4.4 Effect of a third substituent on the benzene ring Page 1 1 1 2 3 5 6 11 13 13 16 16 16 17 18 20 20 21 23 23 25 25 25 27 27 29 29 30 31 32 33 33 35 35 35 39 41 42 4.5 Summary 4.6 Questions and solutions 43 44 Chapter 5: Reactions of arenes and aryl halides 47 47 47 48 50 53 54 55 5.0 Introduction 5.1 Arenes 5.2 Reactions of Arenes 5.3 EAS reactions of aryl halide 5.4 Removal of halide substituent from inactivated aryl halide 5.5 Summary 5.6 Questions and solutions Chapter 6: Phenols 6.0 Introduction 6.1 Physical and chemical properties of phenol 6.2.1 Industrial manufacture of phenol 6.2.2 Laboratory preparation of phenol 6.3 Reactions of phenols 6.3.1 Ether and ester formation 6.3.2 Nitration and sulphonation of phenol 6.3.3 Bromination and acylation of phenol 6.3.4 Kolbe and coupling reactions of phenol 6.4 Uses of Phenols 6.5 Summary 6.6 Questions and solutions Chapter 7: Anilines 7.0 Introduction 7.1 Preparation and properties of aniline 7.2 Reactions of aniline 7.3 Diazonium ion 7.4 Summary 7.5 Questions and solutions Chapter 8: Polynuclear aromatic compounds: Naphthalene 8.0 Introduction 8.1 Synthesis of naphthalene 8.2 Reduction-oxidation reactions of naphthalene 8.3 Orientation and EAS reactions of naphthalene 8.4 Electrophilic aromatic substitution reactions of naphthalene 8.5 Orientation EAS reactions naphthalenes derivatives 8.6 Summary 8.7 Questions and solutions 57 57 57 59 60 61 61 62 62 63 64 64 64 66 66 66 68 70 72 72 74 74 74 75 76 77 79 80 81 Chapter 9: Anthracene and Phenanthrene 9.0 Introduction and preparations 9.1 Oxidation and Reduction reactions 9.2 Diels-Alder reactions 9.3 Halogenation of anthracene and phenanthrene 9.4 Sulphonation of anthracene and phenanthrene 9.5 Alcohols of anthracene 9.6 Positional activity of anthracene 9.7 Anthraquinones 9.8 Summary 9.9 Questions and solutions 83 83 84 84 85 85 86 86 87 88 88 References 90 Practicals 91 91 92 94 96 98 Electrophilic Aromatic Substitution reactions Exp. 1 Preparation of Nitrobenzene Exp. 2 Reduction of Nitrobenzene to Aniline Exp. 3 Nitration of Aniline to p-Nitroaniline Exp. 4 Preparation of Iodobenzene Exp. 5 Preparation of Methyl Orange (p-(p-DimethylAminophenylazo) Benzene Sulphonic acid, Sodiun Salt Exp. 6 Preparation of p-Di-t-Butylbenzene 100 102 CHAPTER 1 STRUCTURE OF BENZENE AND AROMATICITY 1.0 Introduction This chapter will look at detailed scientific arguments that led to structure establishment of benzene, concept of sp2 orbitals and delocalisation of electrons in p orbitals forming pi () electron cloud above and below benzene molecule. Application of Huckel’s rule to benzenoid compounds to establish aromatic and non-aromatic compounds will be discussed. Some important aromatic compounds in natural systems will be mentioned as appreciation of the importance of aromatic compounds. Objectives By the end of this lesson, you should be able to: Discuss the background and arguments that led to the establishment of benzene structure Describe bonding in benzene molecule Explain using resonance, the stability of benzene ring State reasons why benzene is susceptible to electrophiles State the criteria for aromaticity Distinguish aromatic, benzenoid aromatic and non-aromatic compounds using Huckel’s rule State some of the important aromatic compounds in natural systems 1.1 Historical background on benzene Benzene was discovered by Michael Faraday in 1825 from whale oil and reported empirical formula as CH2. However, the correct molecular formula was established in 1834 when Eihartd Mitscherlich synthesized it and was given as C6H6 and this formula elicited many structure suggestions. Compounds like benzene with relatively few hydrogen atoms compared to carbons are usually obtained from natural oils from either plants or animals. Due to their fragrance, they were called aromatic to distinguish them from aliphatic, which have higher hydrogen to carbon ratio obtained from degradation of fats. Figure 1 shows some of the most common benzenoid compounds. or Naphthalene Benzene Anthracene Phenanthrene Triphenylene Pyrene Figure 1. Some common benzenoid compounds 1.2 Structure of benzene There were various suggestions to the structure of benzene that would fit the formula C6H6. Some of such structures are illustrated in Fig. 2 below. 1. 2. 3. CH2 Dewar Structure Kekule Structure 5. 4. Figure 2. Some of the proposed structures of benzene In the course of establishing the actual structure of benzene the following observations were made: 1. Benzene has only one mono-substituted isomer, C6H5Y (Y = Cl, Br, I, NO2 etc). This argument eliminates structures 3 and 5. 2. Benzene yields only three (3) disubstituted isomers, C6H4XX or C6H4XY. (e.g. C6H4Br2 or C6H4BrNO2). Structure 4 of the diyne is eliminated because it would give two isomers. Structure 2 (Dewar) was eliminated because it would give 6 structures! X X Y X Y Y 1,2 1,3 1,4 From these arguments only the Kekule structure remained. He visualized benzene as a dynamic structure where the double bonds are delocalized (π electrons are non-localized). Use of a circle as in the structure on right-hand below indicates that the electrons are delocalized. The bond lengths in benzene are the same of about ≈ 1.39Å. This is established with Xray diffraction techniques. Ethane C-C ≈ 1.54 Å Ethene C=C ≈ 1.34 Å Thus the carbon-carbon bond in benzene is a hybrid of single and double bond. 1.3 Characteristics of benzene It has been established that benzene has very different properties from closely related molecules. A comparison of benzene with cyclohexene and cyclohexadiene is made. Cyclohexene Cyclohexadiene Cyclohexatriene (Benzene) The alkenes undergo electrophilic addition reactions with H2, Cl2, Br2, I2 etc. Benzene on the other hand prefers to undergo electrophilic aromatic substitution (EAS) reactions. The Table below illustrates some other reactions where benzene behaves differently from the other two related compounds. Reagent 1. KMnO4 (cold, dilute) 2. Br2 in CCl4 3. HI 4. H2 in presence of Ni Cyclohexene/ Cyclohexadiene Rapid decolorization Rapid decolorization Rapid addition Reaction 0 Rapid reaction at 25 C and low pressures Thus benzene has some special stability. Benzene No reaction No reaction No reaction No reaction but slow reaction at high temperatures and pressures Heats of hydrogenation Consider the following heats of hydrogenation of benzene and related compounds. Hydrogenation of the three compounds forms cyclohexane. H (Kcal mol-1) H2, Heat 28.6 2H2, Heat 55.4 3H2, Heat 49.8 3H2, Heat 85.8 (calculated) The information can be represented in form of energy diagram. Cyclohexanetriene (Calculated) Cyclohexanediene Benzene Cyclohexene 85.8 Energy 36.0 Resonance Energy (Kcalmol-1) 55.4 49.8 28.6 Progress of the Reaction Figure 3. Energy diagram of benzene and related compounds It is observed that the heat of hydrogenation of benzene is lower than expected by 36.0 Kcal mol-1. This is known as stabilization or resonance energy. This is because the hydrogenation is an addition reaction that converts benzene into a less stable structure as the aromaticity is lost. It is, therefore, easier for benzene to undergo substitution reactions where the aromatic properties are retained. Orbital picture of benzene All carbon atoms of benzene are sp2 hybridized. Therefore, it is a trigonal planer with bond angles of about 1200. It has six (pi) electrons (3 bonds), which are delocalized in the p-orbitals. It is visualized as if there is a cloud of the electrons above and below the plane of the benzene molecule. The electrons are loosely held than the (sigma) electrons and are available for starting a reaction. Hence, benzene is susceptible to electrophilic reagents. E H 2 E+ E + Y 1 H Y2 1 E Stable as the aromaticity is retained Question: Identify the type of reaction indicated by 1 and 2 above and comment on the stability of the compounds. 1.4 Aromaticity and non-aromaticity Aromatic compounds are those that resemble benzene in behavior. They must have the following features: 1. 2. Delocalized electrons. The delocalized electrons that satisfy Huckel’s rule; 4n + 2; where n = 0, 1, 2, 3, 4 etc. That is, the total number of electrons must 2, 6, 8, 10, 14, 18 etc. 3. 4. Cyclic. Planar. Note: There are aromatic compounds that do not resemble benzene in terms of structure and may not contain the phenyl ring. They are mentioned in 1.5.1. The definition of aromatic compounds can therefore be modified to read: Aromatic compounds are those cyclic planar compounds in which the electrons energy of the cyclic form is lower than that of the open chain. 1.5 Benzenoid aromatic and non-benzenoid compounds Benzenoids are compounds, which resemble benzene in terms of structure and behavior. Benzene Naphthalene Benzene has six electrons that are delocalized. It is planar and cyclic. According to Huckel’s rule, it has six pi electrons, Thus, 6 = 4n + 2 n = 1. Therefore it is aromatic. Naphthalene has 10 electrons (5 double bonds) that are delocalized. It is planar and cyclic. 10 = 4n + 2 n = 2. It is aromatic. Anthracene Naphthacene Anthracene has three benzene rings fused in a linear manner. It has 14 electrons (7 double bonds) that are delocalized. It is planar and cyclic. 14 = 4n + 2 n = 3. It is aromatic. Naphthacene (Not naphthalene!) has 18 electrons (9 double bonds) that are delocalized. It is planar and cyclic. 18 = 4n + 2 n = 4. It is aromatic. Triphenylene Triphenylene has 18 electrons (9 double bonds) that are delocalized. It is planar and cyclic. 18 = 4n + 2 n = 4. It is aromatic. 1.5.1 Non-benzenoid aromatic compounds These are compounds that are aromatic but are not benzenoid, that is, they do not resemble benzene and do not contain the phenyl group. - The structures represent cyclopentadienyl anion. As shown the negative charge can be delocalized to other positions in the structure. It has 6 electrons (2 double bonds plus one negative charge) and they are delocalisable, it is planar and cyclic. 6 = 4n + 2 n = 1. It is aromatic. The anion forms stable complexes with transition metals because of aromatic properties. Consider cyclopentadienyl cation. Is it aromatic? + + + The positive charge can be delocalisable; the cation is planar and cyclic. It has 4 electrons; thus 4 = 4n + 2 n = 0.5. This is not an integer and therefore system does not satisfy the Huckel’s rule. Thus the cation is not aromatic. Question: Explain why cyclopentadiene is a stronger acid than most other hydrocarbons. Answer: Acidity is loss of a proton by a given species (like a molecule). Cyclopentadiene therefore dissociates into a hydrogen ion and cyclopentadienyl anion, which has been described as stable because of being aromatic. Therefore the dissociation is favored. This is cycloheptatriene cation formed after loss of a hydride (H-). It has six electrons delocalisable electron (the positive charge be delocalized). It is planar and cyclic. 6 = 4n + 2 n = 1. It is therefore aromatic. The corresponding anion is not aromatic because it does not obey Huckel’s rule. + Cyclopropenyl cation has two electrons which are delocalisable electron. It is planar and cyclic. 2 = 4n + 2 n = 0. It is therefore aromatic. Its corresponding anion is not aromatic because it does not obey Huckel’s rule. Azulene Azulene is a compound containing a seven membered and five membered rings fused together. It has 10 delocalisable electrons. It is planar and cyclic. 10 = 4n + 2 n = 2. It is therefore aromatic. Note: Sometimes delocalized electrons do not have to be the electrons but can be non-bonding electrons (lone pairs of electrons). This is the case in hetero aromatic compounds where the heteroatoms like O, N, S or P are introduced in the ring. Their lone pairs of electrons are able to contribute to the aromatic behavior. Some examples are sited below. HN N H + - N H + Pyrole is a five-membered ring with nitrogen as the heteroatom. By utilization of the lone pair of electrons of nitrogen, there are six delocalisable electrons. It is planar and cyclic. 6 = 4n + 2 n = 1. It is therefore aromatic. :O: :S: Furan and thiophene have oxygen and sulphur as the heteroatoms, respectively. By utilization of one lone pair of electrons of oxygen or sulphur, the molecules have six delocalisable electrons each and will obey Huckel’s rule. .. N Pyridine Pyridine is planar cyclic and obeys Huckel’s rule. Note that the lone pair of electrons in nitrogen is not utilized in this case. It has similar properties as the benzenoids and undergoes electrophilic aromatic substitution reactions. 1.5.2 Anti-aromatic compounds These are compounds in which the cyclic forms have higher electrons energy than the open chain analogues. When the ring opens up what is formed is still anti-aromatic. The best example representing this group of compounds is cyclobutadiene. -H2 Lower Energy Higher Energy -H2 . Lower Energy . Higher Energy -H2 + Lower Energy Higher Energy . -H2 Lower Energy + . Higher Energy All the species do not obey Huckel’s rule and as indicated the cyclic forms are of higher energy then their open analogues. 1.5.3 Benzenoid compounds that do not satisfy Huckel’s rule There are some compounds that contain benzene rings fused in different ways but do not satisfy the Huckel’s rule, the main characteristic feature of aromaticity, yet they are still considered aromatic. Consider the following examples: Pyrene 10b,10c-Dihydropyrene Which of these two compounds is aromatic and which is not? Pyrene is cyclic, has delocalisable electrons and planar. But it has 16 electrons (8 double bonds) and applying Huckel’s rule; 16 = 4n + 2 n = 3.5, it does not satisfy it. However the molecule is considered aromatic. 10b,10c-Dihydropyrene on the other hand has 14 electrons and satisfies Huckel’s rule and all other conditions of aromaticity. It is considered as ‘fully aromatic’ in comparison with pyrene. Benzo[def]chrysene (Benzo[a]pyrene) 12b,12c-Dihydrobenzo[def]chrysene (12b,12c-Dihydrobenzo[a]pyrene) Benzo[a]pyrene has 20 electrons and will therefore not satisfy the Huckel’s rule. But it is planar, has delocalisable electrons and cyclic. It is also considered as aromatic. 12b,12c-dihydrobenzo[a]pyrene with 18 electrons is fully aromatic as it satisfies the Huckel’s rule. Note: Huckel’s rule strictly applies to mono-benzenoid compounds. 1.5.4 Annulenes These are non-benzenoid multi-cyclic compounds. Some of annulenes are not aromatic yet they may be satisfying the Huckel’s rule. Consider [10]annulene for example. Naphthalene Cyclodecapentaene ([10]annulene) A closer look indicate that [10]annulene can be considered to be modified from naphthalene whereby the bridging bond is absent. However, it remains with 10 electrons like naphthalene. It therefore satisfies Huckel’s rule. But it is not planar and not considered as aromatic. Anthracene Cyclotetradecaheptaene ([14]annulene) When anthracene loses the two bridging bonds, it forms [14] annulene, which retains the seven double bonds (14 electrons). Like [10]annulene, [14]annulene is cyclic satisfies the Huckel’s rule and the electrons are delocalisable. Unlike [10]annulene, however, [14]annulene is planar and therefore aromatic. Bicyclo[4.2.0]octa-1(8),2,4,6tetraene Cyclooctatetraene ([8]annulene) Both of the above compounds are not aromatic. Although [8]annulene has the electrons delocalisable and cyclic, is not planar, does not satisfy Huckel’s rule. It is termed as antiaromatic because it is of higher electrons energy than its open analogue, octa-1,3,5,7tetraene. 1.6 Some of aromatic compounds in natural systems Aromatic compounds are of importance in natural systems. Many redox reactions in the cell occur due to the presence of co-enzymes (complex compounds required for any enzyme to function in a biological reactions), which are aromatic in nature. Some of the most important amino acids are aromatic. These are phenylalanine and tyrosine. O O H2N CHC OH H2N CHC OH CH2 CH2 OH 2-Amino-3-phenyl-propionic acid (Phenylalanine) 2-Amino-3-(4-hydroxy-phenyl)-propionic acid (Tyrosine) Structures of a number of hormones contain aromatic rings. A good example is adrenaline, which acts as a transmitter of nerve impulses. O HO NH2 CO2H 2-Amino-3-[4-(4-hydroxy-3,5-diiodo-phenoxy)-3,5-diiodo-phenyl]-propionic acid (Adrenaline) Estrone (oestrone) is produced by mammalian ovaries and controls development of female characteristics and menstruation cycle. As a drug, it is used for replacement therapy in deficiency states like primary amenorrhoea (abnormal absence of menstruation), delayed onset of puberty, control and management of menopausal syndrome and malignant neoplasm of the prostate. O H H H HO Estrone Deoxyribonucleic acid (DNA) and Ribonucleic acid (RNA) are essential for storage of genetic information and synthesis of enzymes and proteins needed for metabolisms. They are composed of, among other chemical features, various aromatic bases of purine and pyrimidine, which are arranged different sequence. O NH2 N N N N N H N Purine N H N N H N H NH2 N O N N Pyrimidine NH Guanine Adenine NH2 N N NH O Cytosine N H O Thymine Note that genetic information flows from DNA to RNA to proteins but in case of retroviruses like HIV the information flows is reverse, that is, RNA to DNA to proteins. Melanine, a polymer of indole derivative, is the dark pigment of the skin. O COOH N H O HOOC O N H O Melanine 1.7 Summary Benzene molecule has increased electron density due to delocalized electrons in sp2 orbitals. Benzene is very stable due to high resonance/delocalisation energy. Aromatic nature of benzene makes it to have unique chemical properties. Anti-aromatic compounds have higher energies in cyclic form than in the corresponding open chain analogues. The conditions have to be satisfied by a compound before it is called aromatic. Benzenoids are aromatic compounds that resemble benzene in terms of structure. There are benzenoid compounds that do not satisfy Huckels rule but are aromatic. Many redox reactions in cells occur due to co-enzymes that are aromatic in nature. 1.8 Questions and Solutions Questions Q1. Q2. (a) Draw all structures that will satisfy the formula C6H6. (b) Assume that mono bromination substitutions were carried out in all the structures in (a) above. Which of the structures will give isomers of the products? Analyze and classify the following chemical species as aromatic, non-aromatic or anti-aromatic. (i) + - (ii) (iii) N (iv) (v) - (vii) (viii) - (vi) (ix) + Solutions 1 (a) 1. 2. 5. 4. (i) (ii) (iii) (iv) (v) CH2 Dewar Structure Kekule Structure (b) 3. Kekule structure(1) has only one product. Dewar structure (2) has two products. Structure 3 has three products. Structure 4 has only one product Structure 5 has two products Br Mono bromination 1 Mono bromination + Br 2 Br Br CH2 Mono bromination Br CH2 + CH2 + CHBr 3 Mono bromination Br 4 Br Mono bromination 5 + Br 2. (i) (ii) (iii) Cycloheptatrienyl cation has 6 electrons, hence, 6 = 4n +2, n = 1. Therefore obeys Huckel’s rule. It is planar, cyclic and electrons are delocalisable. Thus it is aromatic. Cyclopropenyl anion has 4 electrons, hence, 4 = 4n +2, n = 0.5. Therefore does not obey Huckel’s rule. Thus it is non-aromatic. Pyridine has 6 electrons a lone pair on nitrogen which is not involved in delocalisation in this case, hence, 6 = 4n +2, n = 1. Therefore it obeys (iv) (v) (vi) (vii) (viii) (ix) Huckel’s rule. It is planar, cyclic and electrons are delocalisable. Thus it is aromatic. Cyclobutadiene has 4 electrons, hence, 4 = 4n +2, n = 0.5. It does not obey Huckel’s rule. Its open chain analogue has lower electron. Thus it is antiaromatic. Cyclopentadienyl anion has 6 electrons, hence, 6 = 4n +2, n = 1. Therefore it obeys Huckel’s rule. It is planar, cyclic and electrons are delocalisable. Thus it is aromatic. 9bH-Benzopyrene is a poly benzenoid compound. It has 18 electrons, hence, 18 = 4n +2, n = 4. Therefore obeys Huckel’s rule. It is planar, cyclic and electrons are delocalisable. Thus it is aromatic. Cyclooctatetraene has 8 electrons, hence, 8 = 4n +2, n = 1.5. It does not obey Huckel’s rule. Its open chain analogue has lower electron. Thus it is anti-aromatic. Cycloheptatrienyl anion has 8 electrons, hence, 8 = 4n +2, n = 1.5. It does not obey Huckel’s rule. Although its electrons are delocalisable, its cyclic and planar, it is not aromatic due to Huckel’s rule. It is termed as nonaromatic. Cyclopentadienyl cation has 4 electrons, hence, 4 = 4n +2, n = 0.5. It does not obey Huckel’s rule. Its open chain analogue has lower electron. Thus it is anti-aromatic. CHAPTER 2 NOMENCLATURE OF SUBSTITUTED BENZENES AND OTHER BENZENOID COMPOUNDS 2.0 Introduction In chapter one, we looked at the structure of benzene and other benzenoid compounds. In this chapter we are going to look at convectional rules to follow when naming monosubstituted; disubstituted and polysubstituted benzene and other benzene related compounds. IUPAC system of nomenclature will be employed in most cases. However, special names that are internationally accepted will also be used. Objectives By the end of this lesson, you should be able to: State both IUPAC names and special names for monosubstituted benzene Draw structures when given names of monosubstituted benzene State both IUPAC names and special names for substituted benzene Draw structures when given names of substituted benzene State both IUPAC names and special names of other benzene derivatives Draw structures when given names of benzene derivatives State both IUPAC names and special names for substituted naphthalenes and anthracenes Draw structures when given names of substituted naphthalenes and anthracenes 2.1 Nomenclature of monosubstituted benzene Like other organic compounds benzene and other aromatic compounds have various method of naming. However, for continuity IUPAC (systematic) naming is preferred and the following rules will apply. Rule 1. For monosubstituted compounds the name should read in such a way that the prefix of the substituents appear before the word benzene. F Fluorobenzene Cl Chlorobenzene Br Bromobenzene I Iodobenzene NO2 Nitrobenzene Some mono-substituted aromatic compounds have special names that are used instead of the systematic names. NH2 CH3 OH Aniline (Aminobenzene) Toluene (Methylbenzene) Phenol (Hydroxybenzene) COOH SO3H Anisole (Methoxybenzene) CHO COCH3 Benzenesulphonic acid (benzenesulfonic acid) Benzoic acid OCH3 Benzaldehyde Acetophenone 2.2. Nomenclature of substituted benzene Rule 2. If there are several substituents on the ring then the relative positions are shown by numbering the carbon atoms to which they are attached. Numbering starts from the most electronegative group and the sum of the numbers must be the minimum. NO2 m p Note: NO2 o o 1 - 2 ortho (o) 1 - 3 meta (m) 1 - 4 para (p) Cl m Br 3-Bromonitrobenzene (m-Bromonitrobenzene) 2-Chloronitrobenzene (o-Chloronitrobenzene) Rule 3. When a group with special name is present the parent special name is used. CH3 OCH3 NH2 OH HO2C NO2 3-Nitroaniline (m-Nitroaniline) NO2 Br 2-Nitrophenol (o-Nitrophenol) I 2-Iodobenzoic acid SO3H 4-Toluenesulfonic acid 4-Bromoanisole (p-bromoanisole) Rule 4. When two or more similar groups are present then the prefix di, tri, tetra, penta, hexa etc is used to denote the number of groups present. Cl NO2 Br Cl Cl Br O2N Cl 1,2-Dibromobenzene (o-Dibromobenzene) 1,3-Dichlorobenzene (m-Dichlorobenzene) Cl NO2 Cl 1,3,5-Trinitrobenzene 1,2,4,5-tetrachlorobenzene 2.3 Nomenclature of other benzene derivatives Rule 5. If more than two groups that are different are attached to the benzene, the numbering is done from the most electronegative to give the minimum sum possible and prefix must be in alphabetical order. Cl Br NO2 Br Cl I Br NO2 Cl 2-Bromo-3-chloronitrobenzene 5-Bromo-3-chloronitrobenzene 3-Bromo-1-Chloro-5-Iodobenzene Rule 6. If there is a special group, then the derivative is given the special name and numbering started from there. OH NH2 Cl Br Br NO2 O 2N NO2 2-Chloro-4-nitrophenol Br 2,4,6-tribromoaniline CH3 2,6-Dinitrotoluene Rule 7. If there is an alkyl group that is not branched, the benzene nucleus is taken as the parent. CH3 CH2CH3 CH2CH2CH3 Methylbenzene (Toluene) Propylbenzene Ethylbenzene Rule 8. If there are two or more alkyl groups the longer one takes preference. CH2CH3 CH2CH2CH3 3-Ethylpropylbenzene Rule 9. If the alkyl group is branched, the alkyl group is taken as the parent and benzene nucleus as a substituent. 2-Methyl-1-phenylpropane 2-Methyl-2-phenylpropane 3-Phenylheptane Rule 10. If the chain is branched and benzene ring has further substituents on it, then the branched chain still taken the parent. NO2 CH3 2-(4'-Methyl-3'-nitrophenyl)-2,5-dimethylhexane Note: = CH2 C6H5 = Phenyl group = C6H5CH2 = Benzyl group = C6H5CO = Ph O = Benzoyl group = Bz 2.4 Nomenclature of derivatives of naphthalene Unlike benzene the carbon atoms of naphthalene are not equivalent. It has three types of carbon centers as indicated below. The bridging carbon atoms lack the hydrogen atoms and are not numbered, as they cannot be involved in any form of substitution. As the carbon atoms are not equivalent in naphthalene, introduction of a substituent gives two isomers. Before any substituent is introduced both rings are equivalent. For that there are two alcohols of naphthalene and like phenol, they have special names. If substituents are more than one, the numbering follows the order shown above. Examples are shown below. 1 8 7 6 5 a 2 b 3 b b b a 4 a a Fused carbon atoms OH NO2 NH2 OH 1-Naphthol (1-Hydroxynaphthalene) (a-Naphthol) 2-Naphthol (b-naphthol) NO2 2,4-Dinitro-1-naphthalamine OH SO3H NO2 NO2 1,5-Dinitronaphthalene CH3 Br Cl H2N 6-Amino-2-naphthalene sulfonic acid 7-Chloro-1-naphthol 1-Bromo-8-methyl naphthalene 2.5 Nomenclature of anthracene The numbering in anthracene is different from that of benzene and naphthalene in that the middle ring is numbered last. That is, the two carbon atoms of middle ring are numbered 9 and 10. Due to presence of more positions anthracene has very many varied derivatives. A few selected examples are given below. 8 9 1 2 7 3 6 5 10 4 OH Anthracen-9-ol (9-Anthrol) OH OH OH Anthracen-1-ol Anthracene-9,10-diol (1-Anthrol) O O O 9-Anthrone 9,10-Anthraquinone OH NO2 NO2 9,10-Dihydro-anthracen-9-ol 9,10-Dinitroanthracene SO3H SO3H HO3S Anthracene-2-sulfonic acid Anthracene-2,6-disulfonic acid 2.7 Nomenclature of phenanthrene Numbering in phenanthrene is similar to that of anthracene. It is important to note the difference between the two three-ringed aromatic classes of compounds. In anthracene the three ring are fused linearly while in phenanthrene the fusion is not linear but in such a way that the two middle carbon atoms be on the same side. In anthracene there are many positions thus many derivatives. 3 4 2 5 1 6 10 7 8 OH 9 Phenanthren-9-ol Phenanthrene OH O OH O Phenanthrene-9,10-diol Phenanthrene-9,10-dione O 10-Hydrophenanthren-9-one 9,10-Dihydrophenanthrene HO OH Phenanthren-1-ol Phenanthren-4-ol Cl NO2 NO2 1,10-Dinitrophenanthrene Cl 2,7-Dichloro-phenanthrene 2.8 Summary In IUPAC nomenclature of benzenes, the name benzene appears as a suffix if the name is based on benzene. Relative positions; 1:2, 1:3 and 1:4 on benzene ring are referred to as ortho (o), meta (m) and para (p) respectively and sometimes used in nomenclature. Numbering starts at the special group if present on benzene ring and hence special name is used as parent. When branched alkyl group is attached to benzene ring, it is taken as the parent and benzene becomes a substituent. Naphthalene has two different positions while anthracene and phenanthrene have three each. In anthracene and phenanthrene the two carbon atoms of the middle ring is numbered last as positions 9 and 10. 2.9 Questions and Solutions Questions Q1. Give the systematic (IUPAC) names for the following compounds NH2 Br Br COCl CH3 C C (iii) (ii) (i) H3C Cl OH OCH3 (iv) CH2CHCH3 (v) Br O3N NO2 (vi) CO2H NO2 F Br OH (vii) SO3H NO2 (viii) Cl (ix) Cl NO2 OH Q2. Draw structures of the following compounds. (i) (ii) (iii) (iv) (v) (vi) H NO2 4-Cyclohexylanisole 3-methylphenylbenzoate 3-Bromo-5-nitrobenzene sulphonic acid 3-Chloro-7-fluoro-1-naphthol 2-amino-4-butoxybiphenyl 2,7-anthracenedisulphonic acid OH Solutions 1. (i) (ii) (iii) (iv) (v) (vi) (vii) (viii) (ix) 2-Bromo-4-chloroaniline 2-Bromo-5-nitrobenzoyl chloride (cis)-3-Phenyl-2-butene or 3-phenylbut-2(Z)-ene 3-Bromo-5-methoxybenzoic acid 1-Phenyl-2-(3’-fluorocyclopentyl)propane 2,4,6-Trinitrophenol 5-Nitro-2-naphthol 8-Bromo-3-chloro-5-hydroxy-1-naphthalene sulphonic acid 6-Chloro-9-nitro-1-anthrol 2. Structures O (ii) H3C H3CO (i) SO3H (iv) (iii) O H2N C OH Cl Br O2N (v) F O HO3S (vi) SO3H CHAPTER 3 REACTIONS OF BENZENE 3.0.Introduction In chapter one, we looked at what aromaticity is and chapter two, we looked at how various benzene derivatives and benzenoids are named using IUPAC names and some times special names. In this chapter, we will look at influence of aromaticity to reactions shown by benzene. Detailed method for writing the electrophilic aromatic substitution (EAS) reaction mechanisms will be discussed. Halogenation, nitration, nitrosation and Friedel-Crafts reaction will be used to illustrate EAS reactions shown by benzene. Objectives By the end of this lesson, you should be able to: Explain using mechanism, the reduction of benzene. Describe using curly arrows, the general mechanism for electrophilic aromatic substitution (EAS) reaction. Write equations to show the generation of specific nucleophiles for EAS reactions. Write down reaction mechanisms for halogenation, nitration, sulphonation, nitrosation and Friedel-Crafts reactions. Apply EAS patterns to synthesize substituted benzenes. 3.1 Reduction of benzene As had been stated earlier benzene prefers to undergo other reactions where aromaticity is retained. However, under pressure and in presence of a metal catalyst like nickel, platinum or palladium, benzene reacts with three moles of hydrogen to form cyclohexane. H2/Ni High pressure + fast fast slow Benzene H2/Ni H2/Ni Cyclohexadienes Cyclohexene Cyclohexane First step is slow due to aromatic character of benzene as compared to cyclohexadienes. The intermediates in this reaction, cyclohexadienes and cyclohexene, cannot be isolated because they under go reduction very fast under same conditions. Birch Reduction This is a form of limited reduction discovered by Birch, an Australian chemist. The method is for producing 1,3-cyclohexadiene from benzene. Na, NH3, t-BuOH + 1,4-Cyclohexadiene 1,3-Cyclohexadiene Major product (Minor product) The reaction involves the treatment of benzene derivative with sodium (or lithium) metal in liquid ammonia or ethanol. The metal dissolves in the ammonia and so these types of reactions are also referred to as a dissolving metal reduction. Mechanism: Na. - . . B A Note that intermediate B is more stable than intermediate A because of electrons (negative charge and the radical) are furthest from each other hence least repulsion. Each of these intermediates reacts further with ammonia and sodium to form their respective products. H . NH2 A NH2 1,3 Na. H . B - H . NH2 - H . Na. NH2 1,4 Although 1,3 product is stable due to conjugation, 1,4 product is preferred and formed due to its formation from a stable intermediate. 3.2 Electrophilic aromatic substitution (EAS) reactions of benzene: A general mechanism These are the most common reactions that benzene and other aromatic compounds undergo. Due to the electrons cloud the aromatic ring acts as a nucleophile that is attacked by an electrophile. Generally the mechanism involves formation of sigma complexes followed by elimination, which restores aromaticity to the ring. [E+ = Electrophile; B- = Base] H H E E+ + + H E E + Sigma Complexes H Then, E E B- + 3.3 Halogenation Bromination: Bromine molecule itself is not sufficiently reactive to participate in EAS. A Lewis acid catalyst must be added. Br Br2 + HBr + FeBr3 FeBr3 Initial step is formation of a Br2-FeBr3 complex that serves as the E+ in generation mechanism. 2Fe + 3Br2 FeBr3 + Br-Br Br-Br-FeBr3 2FeBr3 Br-Br-FeBr3 Br- FeBr3 H Br + Resonance-stabilized Sigma complex Br + FeBr4+ HBr + FeBr3 Formation of the sigma complex is the rate-determining step. This step is highly endothermic, while the reaction as a whole is exothermic. Br2 H Br FeBr3 + Slow RDS Br Fast Resonance-stabilized Sigma complex Figure 4. Energy diagram of bromination of benzene Chlorination: Chlorination has an analogous mechanism to that of bromination. AlCl3 can be used as the Lewis acid catalyst. Cl2 Cl + HCl + AlCl3 AlCl3 Chlorobenzene Iodination: Requires more specialized conditions. Nitric acid (HNO3) is used as a promoter (not catalyst). Iodonium ion is the electrophile generated with the help of nitric acid as shown below. I I2 NO2 + HNO3 + H 2O Iodobenzene 2H+ + 2HNO3 + I2 2I+ + 2NO2 + 2H2O 3.4 Nitration Direct reaction of nitric acid with benzene is slow. The reaction is sped up by the addition of sulfuric acid. HNO3 NO2 + H 2O H2SO4 Nitric acid and sulfuric acid react to give the nitronium ion, which is the active electrophile (E+). O N+ -O OH O N+ O H O S OH O -O OH2 O N O+ + O O S OH O + H2O Nitronium ion Other nitrating agents are nitronium perchlorate (NO2+ClO4-) and nitronium fluoroborate (NO2+BF4-). 3.5 Sulphonation Reaction of benzene with SO3 in H2SO4 (fuming sulfuric acid) gives benzene sulphonic (sulphonic) acid. SO3 H2SO4 SO3H Benzene sulfonic acid SO3 is a strong electrophile and unlike nitration and halogenation, sulphonation is reversible. O O O S O SO3H O SO + H2SO4 Heat + SO3 3.6 Friedel-Craft alkylation Reaction of aromatic compounds with alkyl halides in presence of Lewis acid, affords alkylated products. R AlX3 + + RX X = Cl, Br, I + HX R = Alkyl group Cl AlCl3 + HCl In the first step the reaction is generation of the carbocation, which is the electrophile. The next step is attack of the electrophile to the aromatic system to give the sigma complexes, followed by elimination. Cl AlCl3 + + H AlCl4- AlCl4- + + Resonance stabilized Sigma complexes For 20 and 30 alkyl halides, the ‘naked’ carbocation is likely involved, as they are relatively stable. The carbocations formed from 10 alkyl halides are much less stable and so a complex between the alkyl halide and Lewis acid is probably the species attacked by the aromatic ring. H AlCl4- CH2CH3 H2C Cl AlCl3 CH2CH3 + CH3 Note: There are three limitations to alkylation reactions. 1). Only works well for benzene, halobenzenes and activated ring systems. 2). Electrophilic species are carbocations that are prone to rearrangement. 3). Multiple alkylations frequently occur because the alkylated product is more activated then benzene itself. H CH3 H C C CH3 AlCl3 + H Not Formed CH3 + Product formed via CH3 H 0 C C CH3 rearrangement of a 2 carbocation 0 CH3 H to a 3 carbocation CH3 H3C C C Cl CH3 H H CH3 H3C C C Cl CH3 H AlCl3 H + H3C C C CH3 CH3 H Rearrangement H + H3C C C CH3 CH3 H 30 carbocation 20 carbocation 3.7 Friedel-Crafts acylation An acyl group has a carbonyl group attached to hydrogen or an alkyl group. The reaction is of acyl halide and a Lewis acid. O + R Cl AlCl3 O R + HCl Lewis acid assists in generation of an ‘acylium ion’ as the electrophile O R O + AlCl3 Cl + + AlCl4- O R R Acylium ion Unlike the Friedel-Crafts alkylation the acylation reaction does not suffer from rearrangement of the electrophile nor is the product susceptible to further reaction. The acylation reaction can be used to synthesize alkyl benzenes indirectly. For example, Clemmensen reduction can be used where carbonyl group is converted to methylene (CH2) groups upon reaction with a zinc/mercury amalgam. O CH3 CH3 Zn (Hg) HCl 3.8. Nitrosation The reaction involving nitrous acid is known as nitrosation. The electrophile involved is nitroso cation generated by nitrous acid. NO + HNO2 + H2O Nitrosobenzene OH- HONO + NO+ H NO + NO+ + OH- NO 3.9 Summary Benzene undergoes mainly EAS reactions to give various substituted products due its aromaticity. Products of EAS of benzene are used as either intermediates or final products or as precursors on industrial scale. Fiedel-Crafts alkylation gives mixed products due to rearrangement of the resulting carbocation. Therefore, it is not a good method to synthesis alkylated benzene derivatives. Fiedel-Crafts acylation does not have rearranged products thus preferred. 3.10 Questions and solutions Questions Q1. (a) By use of reaction mechanisms show how the following transformations are carried: (i) Benzene to nitrobenzene (ii) Benzene to ethylbenzene (iii) Benzene to nitrosobenzene (iv) Benzene to benzene sulphonic acid (v) Benzene to Iodobenzene Q2. Explain the following by use of appropriate structures and/or equations. (i) Friedel-Craft alkylation reactions leads in formation of a mixture of alkylated benzene compounds. (ii) In nitration of benzene concentrated sulphuric acid is required. Solutions 1. Refer to the notes within this chapter. 2.(i) The carbocation formed when the alkyl halide react with a Lewis acid is prone to rearrangement leading to more alkylated products. Consider 1-chloropropane. AlCl3 CH3CH2CH2Cl CH3CH2CH2+ + AlCl4- Rearrangement CH3CHCH3+ a more stable carbocation These two carbocations will react with benzene to give two different products although the secondary carbocation will form the major product, isopropylbenzene. That was not the initially expected product. H3C C CH3 CH2CH2CH3 Propylbenzene Minor Product H 2-Phenylpropane (Isoprylbenzene) Major Product It is also noted that the alkylated product formed is activated to electrophilic substitution reactions than benzene thus forms multi-alkylated benzene products at ortho and para positions. R R R R R R R R R R R CHAPTER 4 REACTIONS OF BENZENE DERIVATIVES 4.0 Introduction In chapter three we looked at EAS reactions of benzene with various nucleophilic reagents. In this chapter we are going to study the effect of groups attached to benzene ring in terms of how they affect rate of electrophilic aromatic substitution (EAS) reactions and position at which the incoming substituent gets attached to the ring. These reactions will also be used to show the synthesis of various substituted benzenes. Industrially useful substituted benzenes will also be discussed. Objectives By the end of this lesson, you should be able to: Explain the effect of electron donating group on benzene on the rate of EAS and orientation of an in coming nucleophile Explain the effect of electron withdrawing group on benzene on the rate of EAS and orientation of in coming nucleophile Describe how the rate of EAS reaction of benzene and substituted benzene is determined State characteristics of common ortho-para and meta directors Explain using resonance structures, the effect of a group on benzene that donates electrons by induction and that releases electrons by resonance on EAS Explain why halogens are deactivators yet they are ortho-para directors to an incoming electrophile Explain the effect of two substituents on benzene to the rate reaction and orientation of incoming substituent State some of industrially useful substituted benzenes 4.1 Effect of substituents on EAS patterns and orientation 4.1.1 Nitration of Toluene Like benzene, toluene undergoes electrophilic aromatic substitution reactions. The reaction is faster than that of benzene. Nitration reaction, for example, is about 25 fold faster. The reaction gives three products, two major and one minor and almost not realized. CH3 CH3 CH3 CH3 HNO3 + + H2SO4 NO2 NO2 o-Nitrotoluene 40% m-Nitrotoluene 3% O2N p-Nitrotoluene 57% If nitration were random, a 20:40:40 para:meta:ortho ratio would be produced. This is due to the fact that there are two ortho positions, two meta positions and one para position. However the situation is not observed experimentally. The substituents have effects on EAS of benzene derivatives. In the above case of nitration of toluene the ortho and para products are favored. From the above observations on nitration of toluene two conclusions can be made about the methyl group attached to benzene ring: 1. It is an activating group because the reaction is faster than that of benzene towards electrophilic substitution. 2. It is an ortho–para director. Recall that the rate-determining step is the formation of the sigma complexes. The complexes leading to the ortho and para products are more stable than that leading to the meta product. Accordingly, the transition states leading to the ortho and para products are of lower energy than those leading to the meta product. CH3 Ortho CH3 + + H NO2 CH3 H NO2 + H NO2 Two with 20 carbocations, one with a 30 carbocation. CH3 H2SO4 + CH3 HNO3 Meta CH3 CH3 + + H NO2 H NO2 H NO2 All three with 20 carbocations CH3 Para CH3 + + H O2N + H H O 2N O2N Two with 20 carbocations, one with a 30 carbocation. CH3 Figure 5. Energy diagram of nitration of toluene 4.1.2 Nitration of nitrobenzene Unlike the case of nitration of toluene, nitration of nitrobenzene is a very slow reaction. It is about 105 times less reactive to EAS reactions than benzene. The reaction takes place at higher temperature when compared with the nitration of benzene. The reaction leads to formation of three products, with m-dinitrobenzene, the meta product predominates and para product almost not realized. NO2 NO2 NO2 NO2 HNO3 + + H2SO4 1000C NO2 o-Dinitrobenzene 6% NO2 m-Dinitrobenzene 90% O 2N p-Dinitrobenzene 0.7% The nitro group is electron withdrawing which deactivates the ring towards EAS. The group removes electron density from the ring and slows down the reaction. O O- N + O- N + O From the above observations on nitration of nitrobenzene two conclusions can be drawn about the a nitro group attached to benzene ring: 1. It is a deactivating group because the reaction is slower than that of benzene towards electrophilic substitution. 2. It is a meta director The sigma complex leading to the meta product is less destabilized than those leading to the ortho and para products. O +N O NO2 + + NO2 Ortho H NO2 H NO2 + H NO2 Especially destabilized (Adjacent + charges) NO2 H2SO4 + NO2 HNO3 Meta NO2 NO2 + + H NO2 H NO2 NO2 Para H O2N + + H NO2 O N O + + H H O 2N O2N NO2 Especially destabilized (Adjacent + charges) Generally groups that deactivate the ring are usually meta directors. Other deactivating meta directing groups include, ketones, esters, nitriles, sulphonic acids and ammonium salts. All are either positively charged, or have a resonance form in which a positive charge is immediately adjacent to the ring. 4.1.3 Determination of relative reactivity of reaction 1. Time required for a reaction to occur under identical conditions 2. The severity of conditions (temperature, pressure and concentration) required for a comparable reaction to occur under other identical conditions. For example, nitration of benzene and nitrobenzene for one hour is at 600C and 900C, respectively. Benzene is therefore more reactive than nitrobenzene. 3. Competitive reactions whereby a mixture of equal moles of compounds is made to compete for a limited amount of reagent and quantitative analysis of the product and the reactants done. 4.1.4. Orientation of the nitro group The table below presents some information on the nitration of monosubstituted benzene. From the table below one can be able to come up with a classification of the groups as ortho-para directors or meta directors. Group attached to benzene -OH -CH3 -OCH3 -NH2 -CH2CH3 -Cl -Br -NHCOCH3 -NO2 -CO2H -CN -SO3H -CHO -+N(CH3)3 Relative percentage of product Ortho Meta Para 55 40 45 50 35 35 40 19 6 20 21 0 0 3 1 0 1 1 0 2 90 80 81 72 72 89 45 57 55 50 63 64 60 79 1 0 7 11 Strong activating agents (ortho-para directing): -NH2, -NHR, -NR2, -OH, OMe Moderately activating (ortho-para directing): -NHCOCH3 Weakly activating (ortho-para directing): -C6H5, -CH3, -CH2CH3 Deactivating agents (ortho-para directing): Halogens (-F, -Cl, -Br, I) Deactivating agents (meta directing): -NO2, -CO2H, -CN, -SO3H, -CHO etc Note: a) An activating group activates all the positions on the benzene ring including the meta position but activates ortho and para positions more than meta. b) A deactivating group deactivates all the positions on the benzene ring including the meta positions but less than the ortho and para. 4.2 Groups that donate electrons to the benzene There are two types of groups under this category. There are those groups that donate electrons by inductive effect and those that donate by resonance. Groups that donate electrons by inductive effect are groups that do not have lone pair of electrons to donate for resonance stabilization. But they are electron releasing by inductive effect. Thus they increase the electron density to the benzene ring and hence the ring becomes more attractive towards an incoming electrophile than benzene itself. They are however termed weak activators. Alky group, methyl, ethyl, propyl etc, represent this class (refer to nitration of toluene). Groups that release electrons by resonance have lone pairs of electrons, and are powerful activators of ortho and para positions. They include amino (-NH2), and hydroxyl (-OH) groups. They release electrons by means of resonance. Consider a case of nitration of aniline. NH2+ H .. NH2 NH2 + + Ortho NO2 H NO2 NH2 H NO2 + H NO2 NH2 H2SO4 + NH2 HNO3 Meta NH2 NH2 + + H NO2 H NO2 .. NH2 NH2 Para H NO2 + + H O2N + H H O 2N O2N NH2 + NH2 H O2N The lone pair of electrons of nitrogen provides an additional stabilization of the sigma complex by resonance donation of the electrons. There is an additional resonance form for the complexes leading to the ortho and para products. Hence these sigma complexes are more stable. Under some conditions aniline is so reactive that it undergoes EAS without a catalyst. Bromination occurs without the Lewis acid and substitution can takes place in three positions. NH2 NH2 3Br2 Br Br H2O, NaHCO3 Br Sodium bicarbonate is required to react with the HBr that forms. Otherwise, the basic amino group protonates and EAS slow down. Protonated amino group is an electronwithdrawing group and removes electrons density from the ring and slows down the reaction. + NH3 Br- NH2 HBr 4.3 Groups that withdraw electrons from the benzene ring by induction and release electrons by resonance They display two opposing effects: 1. They are electronegative and hence deactivate the ring through inductive effects. 2. They can donate electron density through resonance and hence activate the ring through resonance through formation of a ‘halonium ion’ (C=X+). Net effect is that the rate of EAS is slower for halobenzene than benzene, but these groups give ortho and para products, as the major ones. Br Br HNO3/H2SO4 Br NO2 + + Br O2N NO2 ortho ~35% meta ~1% para ~64% It is observed that like the sigma complexes of aniline, chlorine atom can contribute a lone pair of electrons and the sigma complexes for ortho and para increased by one each through formation of the ‘halonium ion’. + Cl + Ortho .. Cl :.. H NO2 Cl + H NO2 Cl H NO2 + H NO2 Cl H2SO4 + Cl HNO3 Meta Cl Cl + + H NO2 H NO2 Cl Para H O2N + + H NO2 .. Cl :.. + Cl H H O2N O2N + Cl H O2N 4.4 Effect of a third substituent on the benzene ring Presence of already two substituents on the benzene ring complicate the rules mentioned earlier if the third substituent is to be introduced. However, a few general rules for some cases can be predicted by combining the effects of each of the two groups. If the two substituents direct an incoming group to the same position(s), then that will be the principal position of the third substituent. That is, the two substituents may be located in such a way that their directive influence reinforces each other. A few examples are cited below. SO3H CH3 NHCOCH3 H3C NO2 CN NO2 CH3 Highly deactivated Predicted site if EAS works at all Hindered Reaction between two substituents generally not favored due to steric hindrance. Although the two methyl groups in m-xylene would reinforce each other no reaction takes place for the ortho position between the two groups. If two deactivating groups are present, regardless of their position, it may be difficult to effect a third substitution (case of m-nitrobenzene sulphonic acid above). If two groups conflict in their directive effects, the more powerful activator will exert the predominant effect (NH2>OH>OCH3>NHCOCH3>C6H5>CH3>halogens>meta directors. X X2 HO Cl HO Cl H3C NHCOCH3 Br Br2/FeBr3 H3C Major NHCOCH3 H3C NHCOCH3 Minor Br 4.5 Summary Rate of EAS reaction of monosubstituted benzene with an incoming nucleophile and the orientation is affected by nature of the substituent already on the ring. Electron withdrawing groups (activators) are meta directors. Electron donating groups (deactivators) are ortho and para directors. Halogens are ortho and para director although they are deactivators. The position of the third incoming substituent on benzene ring does not follow the usual EAS patterns. 4.6 Questions and solutions Questions Q1. Explain the following by use of reaction mechanisms and resonance structures. (a) A keto group attached to benzene ring is deactivating and directs the incoming electrophile to the meta. Birch reduction of benzoic acid (A) gives compound (B) and not compound (C). (b) CO2H CO2H CO2H B C Li, NH3 (l) EtOH A Q2. Suggest the major organic compounds (D-H) in the following reactions. NHCOCH3 OH Br2, CH3CO2H (i) HNO3 (ii) D E H3C NH2 (iii) Cl2/H2O F NO2 (iv) HNO3/H2SO4 NO2 G CH3 O OCH3 + (v) O O H AlCl3 Solutions 1. (a) The carbon of the carbonyl group is polarized in such a way that the carbon atom of the carbonyl carries a partial positive charge while the oxygen carries the partial negative charge. That is, carbon is in need of electrons. These electrons are supplied by the benzene ring. As the electron density of the benzene is reduced, the carbonyl group is therefore termed as a deactivating group because the benzene ring reactivity towards electrophilic attack has been lowered. O R C R Any electrophilic attack, which would lead to resonance structures whereby the benzene carbon attached to the carbonyl group carbon carries a positive charge, is unfavorable. This is noted in the ortho and para electrophilic attacks. Although the whole ring is deactivated, ortho and para position are more deactivated than the meta where the positive charge does not fall on the carbon atom of the benzene attached directly to the carbonyl carbon as observed in the resonance structures below. COCH3 COCH3 COCH3 COCH3 + HNO3/H2SO4 meta attack + H H H NO2 NO2 NO2 + COCH3 COCH3 HNO3/H2SO4 H NO2 H H ortho attack COCH3 COCH3 COCH3 HNO3/H2SO4 COCH3 + + para attack H + NO2 H NO2 H NO2 1 (b). A carboxylic group is an electron-withdrawing group as the carboxyl carbon highly deprived electrons by the two oxygens attached. The mechanism below will help to explain why the product is formed. CO2H HO C O O- HO EtOH - Li + etc . . Negative charge resonance stabilized by the electron deficient CO2H group HO C O HO O CO2H Li, EtOH . + etc . Favored product Lower energy pathway CO2H CO2H - Li CO2H CO2H EtOH - . . CO2H . Li EtOH Negative charge not stabilized by the CO2H group. (Higher energy pathway) -Not fovorable 2. NHCOCH3 NH2 OH (i) D = Cl (iii) F = (ii) E = NO2 H3C Br NO2 NO2 (iv) OCH3 NO2 G= (v) CH3 H= O C CH2CH2CO2H CHAPTER 5 ARENES AND ARYL HALIDES 5.0 Introduction In chapter four, we looked at EAS reactions of benzene and its derivatives and effect of substituents on rates of EAS reactions as well as their orientation patterns. In this chapter, preparation of arenes and reactions shown by arenes will be studied. Discussion of EAS reaction of aryl halides will be done in addition to those reactions that will lead to removal of the halogen from the ring. Reactions involving aryl halides useful in the synthesis of important compounds will also be mentioned Objectives By the end of this lesson, you should be able to: Describe using equations preparation of some common arenes. Explain with the help of mechanisms why different products are obtained when arenes are halogenated in dark and in presence of light. Predict reaction products when unsaturated arenes undergo halogenation in the presence of Lewis acids. Write down oxidation and reduction products of styrene. Explain deactivating effect of halogens using resonance structures. Describe EAS reactions of aryl halides. Describe how a halogen substituent is removed from activated and inactivated aryl halide. State some of the important arenes, aryl halides and compounds obtained from them. 5.1 Arenes Arenes are aliphatic aromatic hydrocarbons. Recall the methods of naming arenes (chapter 2) and methods of preparations. Note that if a double is present, depending on the position, geometrical isomerism may be exhibited. H H CH3 CH2 H H 3-Phenyl-1-propene H (Trans)-1-Phenylpropene H3C H H (Cis)-1-Phenylpropene Arenes are of low polarity and insoluble in water but soluble in organic solvents like CCl4, ether, hexane etc. Disubstituted para isomers have higher melting point due to better packing. 5.2 Reactions of Arenes Arenes undergo most of the reactions of benzene ring, that is, EAS reactions. They can also undergo reactions involving the side chain. Recall that they are ortho-para directors and activates the ring by inductive effect. The para product usually predominates over the ortho product due to steric effects. Note that the halogenation with Lewis acid is carried out in darkness. In presence of light the side chain will react with the halogen. CH3 CH2CH3 Br2/Light CH2CH3 Br H CH2CH3 CH2CH3 Br2/FeCl3 + Dark Br Br Major Minor 5.2.1. Halogenation of Arenes Halogenation of the side chain is a radical mediated reaction. 1-Bromo-1-phenylethane, shown above is the only product due to stability of the intermediate radical involved being stabilized by the phenyl group by resonance. This is a reactivity selectivity principle. hv Br2 CH2CH3 CHCH3 Br . Br . + . CHCH3 + Br. . + Br2 HBr + CH3 C H Br + Br . Only product Chlorination of ethylbenzene, however, gives two products with 1-chloro-1-phenylethane (91%) predominating over 1-chloro-2-phenylethane (9%). Question: What are the terminating products of the halogenation of ethylbenzene in presence of light? Unsaturated arenes undergo halogenation in absence of the Lewis acid just like alkenes. Br2 H Br Br 1,2-Dibromo-1-phenylethane H H Br2/FeCl3 + Br Br Bromostyrenes major H HBr CH3 Br 1-Bromo-1-phenylethane (Markovnikov's product) Styrene H HBr CH2Br PhCOOOCOPh H 1-Bromo-2-phenylethane (Anti-Markovnikov's product) (Dibenzoylperoxide) 5.2.2 Reduction reactions of arenes The reaction of styrene can occur both on the side chain and also on the ring. Consider reduction with hydrogen in presence of nickel. CH2CH3 H2/Ni Ethylbenzene 200C, 2-3 Atm CH2CH3 H2/Ni Styrene Ethylcyclohexane 1150C, 110 Atm In presence of hydrogen peroxide in acetic acid, styrene is converted to a glycol, which is oxidized further by oxidative cleavage to benzoic acid. Any side chain with the carbon atom that is directly attached to the ring (benzylic carbon), and has at least two hydrogens, is oxidatively cleaved by strong oxidizing agent to benzoic acid. H H2O2/HCO2H OH CH2OH KMnO4 CO2H Styrene Phenyl-1,2-ethanediol (a Glycol) Benzoic acid CO2H KMnO4, OH, HeatH+ CO2H CO2H KMnO4, OH, HeatH+ No reaction Question: How are (i) saturated and (ii) unsaturated arenes prepared? For the saturated arenes refer to Friedel-Crafts alkylation. Recall that the method has its limitations. It was also mentioned that the limitations could be avoided by use of FriedelCrafts acylation later reducing the acylated product by Clemmensen reduction (HCl in presence of Zn/Hg) or Wolf-Krisher reduction (N2H4 in presence of a base). Unsaturated arenes are prepared by normal reduction of acylated products. Note that if the alkyl halides have more than one halogen atom, more than one phenyl groups will be present in the products. 3 2 + CH2Cl2 3 + CHCl3 + CHCl4 AlCl3 Diphenylmethane AlCl3 H Triphenylmethane AlCl3 Cl Chlorotriphenylmethane Triphenylmethane is acidic due to resonance stabilization of carboanion. Use of carbon tetrachloride does not lead to formation of tetraphenylmethane due to steric factors. 5.3 Aryl halides and nucleophilic substitution reactions Aryl halides, as had been mentioned earlier, are compounds in which halogen atom is directly attached to the aromatic ring. Their preparation methods were mentioned in chapter 3. Aryl halides behave differently from alkyl halides. They do not undergo nucleophilic substitution that easily as do the alkyl halides. They do not give a positive silver nitrate test as alkyl halides do. In some case they behave like vinyl halides (a halogen attached to a carbon atom with a double bond), which do not react with nucleophiles. X CH2X or ArX Not an aryl halide Aryl halide R-X + Nu - ArX + Nu - SN1 or SN2 R-Nu + X- No reaction This behavior of aryl halide is due to the contribution of one of the lone pairs of the halogen towards extension of delocalisation. .. :X : .. :X + .. :X + .. :X + - - Thus the ring as a whole is deactivated towards nucleophilic attack. However, in presence of an electron withdrawing group at ortho or para (or both) with respect to the halide activate such a reaction. This is an addition-elimination reaction. Presence of the deactivating group at meta has no such effect on the aryl halide. Cl ON+ O + Cl Nu - Cl ON+ O- ortho Nu Nu ON+ O- Cl Cl Cl + Nu - Nu NO2 Nu para O N+ O -O N+ O - -O N+ O - NO2 - The more the electron withdrawing groups there are at ortho and para the easier it is to replace the halide. Only effected at ortho and para positions. Ring activators cannot effect the change. The order of reactivity of aryl halides to nucleophilic substitution is: I > Br > Cl >F Cl OH NO2 NO2 NaOH, 1300C Reflux OH Cl NO2 NO2 aq NaHCO3 1000C NO2 NO2 OH Cl O2N O2N NO2 NO2 aq NaHCO3 350C NO2 NO2 A variety of products can be prepared depending on the type of substituted aryl halide and the base used. Br 1000C O2N OCH3 CH3ONa O 2N Cl NHCH3 NO2 NH2CH3 NO2 1600C I NC CN NHC6H5 C6H5NH2 EtOH, 950C NC CN Other important reactions involving aryl halides are shown below. Cl CN NO2 CO2H NO2 KCN CO2R NO2 aq H+ CO2R F CO2R Li H 2O + Li Biphenyl NaNH2/NH3 OCH3 OCH3 Br H 2N 2-Amino-4-methoxybiphenyl (4-Methoxy-2-phenylaniline) Br + MgBr THF or Et2O Mg(s) Dry Grignard Reagent 5.4 Removal of halide substituent from inactivated aryl halide For inactivated aryl halides, the substitution is carried out at high temperatures in presence of a strong base. The nucleophilic substitution occurs but under a different mechanism. In the mechanism a benzyne (a benzene a triple bond) is implicated (See example and the mechanism in the page that follows). This reaction cannot take place if there is no ortho hydrogen to be lost in the process of formation of the extra bond. OH ONa Cl H+ H2 O + NaOH High pressure Dow Process NH2 Br + KNH2 + -330C NH3 Cl NH2 NaNH2/NH3 H3C H3C 50% KBr + H 3C 50% NH2 Mechanism: Br H NH2- - NH2- CH3 H NH2 + NH2- NH2 H3C CH3 Benzyne intermediate NH2 CH3 Addition of NH2 to the other end of triple bond provides the other regioisomer Cl H3CO CH3 NaNH2/NH3 No reaction CF3 CF3 Cl NaNH2/NH3 NH2 The last transformation can only be explained by considering a benzyne intermediate. The benzyne generated as the intermediate can be trapped in a Diels Alder reaction. + Benzyne O Furan O Diels Alder Adduct 5.5 Summary Arenes give different products depending on whether the reaction is done in presence or absence of light. Unsaturated arenas undergo halogenation in absence of Lewis acids just like alkenes. Arenes are prepared by Frediel-Crafts acylation of benzene followed by reduction. Halogen substituent on benzene ring deactivates it towards nucleophiles but it is orthopara director. Electron withdrawing groups at ortho and para position on aryl halide makes it easier to remove the halogen from the ring. For inactivated aryl halide, the halogen can be removed via benzyne intermediate at high temperatures. A halogen on the ring is a deactivator but ortho-para director. Aryl halides are very important precursor for synthesizing many benzene derivatives. 5.6 Questions and solutions Questions Q1. With the aid of reaction mechanisms, explain the following observation. Reaction of ethylbenzene (1) with bromine in presence of light gives compound 2 as the major product and compound 3 as the minor product. CH2CH3 CHBrCH3 CH2CH2Br Br2/ light + 1 Q2. 2 3 Given the following reaction, CF3 NaNH2/ NH3 Cl CF3 NH2 (i) (ii) Write a reaction mechanism leading to the above product. Why is the ortho substituted product not formed? Q3. What is the product in the following transformation? Br NO2 Na OCH3+NO2 Solutions 1. See page 53. 2. (a) The reaction involves a benzyne intermediate. Attack of the benzyne by amide ion would lead to formation of two negatively charged benzenes (see below). The compound formed is governed by the stability of one of these carboanions. Note that CF3 group is an electron-withdrawing group, and thus the closer the negative charge to it the better-stabilized carbocation. That inductive withdrawal of electrons (not resonance) by the CF3 group more favored if the negative charge is nearer. This leads to the most favored product (low energy pathway) CF3 Cl NH2- CF3 CF3 Cl Cl- CF3 CF3 NH2 NH2- - - - + NH2 Benzyne intermediate A B NH3 CF3 CF3 NH2 NH2 (b) The ortho product is not formed because the negative charge of the intermediate (B) is far away from the CF3 group thus less stabilized. Therefore less favored product due to high energy pathway. 3. The reaction is a nucleophilic substitution reaction, just like the reaction in 2 above. The nucleophile is OCH3 ion, which replaces the chlorine atom, thus lost as a chloride. The product is 2,4-dinitroanisole. OCH3 NO2 NO2 CHAPTER 6 PHENOLS 6.0 Introduction In chapter five, we looked at arenes and aryl halides, their reactions and importance as precursors for variety of industrially useful organic compounds. In this chapter, phenols, that form the most important derivatives of benzene, will be studied. Physical and chemical properties of phenols that make then unique will be dealt with in details. Illustration of synthesis of compounds utilizing phenols will also be studied. Objectives By the end of this lesson, you should be able to: Explain the physical and chemical properties of phenols. Describe laboratory preparation and industrial manufacture of phenol. Write resonance structures to illustrate ortho-para directing effect of hydroxysubstituent on benzene. Predict reaction under different conditions products of phenols State some of the uses of phenols and their derivatives. 6.1 Physical properties of phenols Phenols are aromatic alcohols in which hydroxyl group is directly attached to the aromatic nucleus. Phenols resemble aliphatic alcohols in many ways. However, there are some case whereby there are distinctive differences between the two hydroxyl containing compounds. Simple phenols are liquids with high boiling points attributed to powerful hydrogen bonding. Normally colorless unless oxidized to quinones, which is responsible for the color of phenol when impure. H O OO N+ O N+ OHO o-Nitrophenol ON O+ H O p-Nitrophenol Ortho-Nitrophenol has intramolecular hydrogen bonding which lowers its solubility in water when compared with para-nitrophenol. Generally, the ortho-nitrophenol has lower boiling point and melting point than meta and para isomers as the intramolecular hydrogen bonding reduces the intermolecular ones. For this, o-nitrophenol can be steam distilled while meta and para isomers cannot be steam distilled easily. Phenols are weakly acidic due to resonance stabilization of the phenoxide ion. This makes them stronger acids when compared with the aliphatic alcohols. Therefore phenol reacts with strong bases like sodium hydroxide but not with weak ones like bicarbonates. Alcohols do react with neither weak nor strong bases. This property can be used to identify and separate phenols from acids and alcohols. O- OH O - B- O O - Presence of electron withdrawing groups at ortho and para positions (but not meta) increase the acidity of phenol. The groups stabilize the resulting phenoxide ion by sharing some of the negative charge by resonance. The more the groups there are in a phenol molecule the stronger the acid. O- O N+ O- OO N +O- O- - O N + O O - O N+ O- Phenol salts are soluble in water. Due to the resonance stabilization of the negative charge on the phenoxide ion it is a poor nucleophile, but a strong base. Since phenolic moiety is common to all structures, the effect of electron delocalisation is the same. Thus, the other factor in operation is either ability to donate electrons to the ring or withdraw electrons from the ring. The following substituted phenols are used to explain the effect: OH pH pH OH OH OCH3 CH3 9.95 10.30 10.19 OH OH OH Cl CHO NO2 7.14 7.66 9.38 From the above phenols, electron-donating groups (OCH3, CH3 etc) reduces the acidity of phenols. This can be explained in terms of how hard it is to break the O-H bond so that H+ is released. Since these groups increase the electron density of oxygen, it is hard for the O-H bond to break or ionize. On the other hand, electron withdrawing groups weaken the O-H bond by reducing the electron density of oxygen and hence easy to break or ionize. The same effect of substitution on pka values are also seen in substituted benzoic acids and substituted protonated anilines. Note: There is a relationship between the pka values and reactivity of benzene ring towards EAS; the more strongly deactivating the substituent is the lower the pka values. 6.2.1 Industrial manufacture of phenol There are two main processes that are used in industries in manufacture of phenol normally for industrial use. Dow process, already mentioned earlier, involves alkyl halide, chlorobenzene heated at 3500C and high pressures in presence of sodium hydroxide. It is first converted to sodium phenoxide, which hydrolysis in presence of an acid gives phenol. OH ONa Cl + NaOH 3500C, H2O High pressure H+ The second method involves cumene (isopropylbenzene) as the raw material. Cumene is air oxidized to form the corresponding organic peroxide, which on hydrolysis in presence of an acid lead to formation of phenol. O CH3 O2 H2O/H+ H3C C OOH Air oxidation CH(CH3)2 CH3 OH Peroxide Cumene + H 3C Acetone Phenol 6.2.2 Laboratory preparation of phenol Various methods can be used to prepare phenols in the laboratory. 1. Hydrolysis of diazonium salts N2+HSO4 OH H2O + H2SO4 + N2 Benzene diazonium bisulphate Question: How can you prepare phenol from benzene, through nitrobenzene? 2. Alkali fusion of sulphonates SO3H NaOH, H2O OH 3000C Benzene sulphonic acid 3. Oxidation of aryl thallium compounds (high yield) Tl(O2CCF3)2 OCCF3 O Pb(OAc)4 (CF3COO)2Tl Ph3P OH H+ O- H2O/OH- 4. Basic hydrolysis of aryl halides (works only if there are electron-withdrawing groups attached at ortho or para with respect to the halide. (Refer to the reactions of aryl halides). 6.3 Reactions of phenols Acidity: As mentioned earlier phenols are acidic in nature and will react with strong bases like sodium hydroxide to form phenoxide salt and water. They partially dissociate in water. OH O-Na+ + NaOH + H 2O Sodium phenoxide O- OH + + H2 O H 3O + 6.3.1 Ether and ester formation Phenols and their salt form ethers when reacted with alkyl halides (William’s synthesis). ArOH ArO-Na+ H3C + ArOCH2CH3 CH3CH2I + ArOR RX OH + BrH2C + NO2 NaX H3C OCH2 NO2 Phenols like aliphatic alcohols react with carboxylic acids and their derivatives like acyl halides and acid anhydrides to form esters. The reaction with the acid is highly reversible and thus not preferred in ester formation. RCOCl OH OCOR or (RCO)2O RSO2Cl OSO2R 6.3.2 Nitration and sulphonation of phenol Phenol will react with nitric acid to form o-nitrophenol and p-nitrophenol. Sulphuric acid is not needed in this reaction as the hydroxyl group highly activates the ring for EAS reactions. The ortho isomer is form in a slightly larger proportion molecules are stabilized by intramolecular hydrogen bonding. OH OH HNO3 OH + NO2 O2N Phenol reacts with sulphuric acid to form two products depending on the temperature under which the reaction is carried out. At low temperatures o-hydroxybenzene sulphonic acid is form. This product is termed as a kinetic product. At higher temperatures, on the other hand, p- hydroxybenzene sulphonic acid is the major product. This product is termed as a thermodynamic product. OH 0 SO3H 15-20 C Kinetic product OH o-Hydroxybenzene sulphonic acid H2SO4 OH 0 100 C Thermodynamic product SO3H p-Hydroxybenzene sulphonic acid 6.3.3 Bromination and acylation of phenol Benzene ring is highly activated by the hydroxyl group such that bromination reaction takes place in absence of the Lewis acid catalyst. Three positions of the ring are substituted when bromine is introduced in presence of water. However, mono bromination at para position when bromine is accompanied by sulphur carbide (CS2) at 00C. OH OH Br Br2, H2O Br 2,4,6-Tribromophenol Br OH OH Br2, CS2 p-Bromophenol Br Phenol and its derivative can undergo acylation in presence of acid anhydride. In presence of a Lewis acid the acylated product undergoes a rearrangement where the acyl group migrates to the ortho or para position depending on the reaction temperature. At room temperature (250C) the group migrates to the para position with respect to the initial occupied position. With temperatures of around 1600C, the ortho isomer is formed. This rearrangement is known as Fries rearrangement. OH O OH AlCl3 OCCH3 (CH3CO)2O CH3 COCH3 250C CH3 CH3 AlCl3 OH 1600C H3COC CH3 6.3.4 Kolbe and coupling reactions of phenol Phenol is converted to ortho-hydroxybenzoic acid when it is treated with sodium hydroxide and later with carbon dioxide. OH ONa NaOH OH CO2 1250C, 4-7 Atm H+ CO2Na Phenols couple with diazonium salt to form azo compounds. They are colored compounds that are used as dyestuff pigments. OH + N+2 N N HO p-Hydroxyazobenzene OH CO2H 6.4 Uses of Phenols 1. Phenol itself is used as antiseptic (antimicrobiol). Modern antiseptics still contain phenolic groups. 2. Phenol formaldehyde resin 3. Manufacture of dyes 4. Additive for odor and flavorings OH OH Cl Cl OH HO Cl Cl Cl CH2(CH2)4CH3 Cl Hexachlorophene n-Hexylresorciol Phenolic Antiseptics Some naturally occurring phenols have been used for a long time in food industries because of their aroma and taste, like thymol (2-isopropyl-5-methylphenol) and vanillin (4-hydroxy-3-methoxybenzaldehyde) from thyme and vanilla beans, respectively. 6.5. Summary Phenols react just like aliphatic alcohols. Phenol undergoes EAS reactions and is an ortho-para director. Phenol is a precursor for many industrially useful compounds such as aspirin. This makes it one of the most important derivatives of benzene. 6.6 Questions and solutions Questions Q1. By use of resonance structures and reaction mechanisms, explain why presence of an electron withdrawing group at ortho and para positions increase the acidic character of phenols. Q2. Outline synthesis of 2,6-Dichlorophenol from phenol Q3. Suggest the major organic compounds (A-C) in the following transformations. OH 1). CHCl3, OH- (i) A 2). H2O/H+ OH NO2 H2SO4, H2O (ii) B Heat SO3H OH NH2 (iii) 1). NaNO2, HCl C 2). CuCl, Heat Br Solutions 1. See page 59 2. OH OH Conc. H2SO4 OH Cl2 1000C Cl OH Cl 1). dil. H2SO4 Cl Heat FeCl3 SO3H SO3H 2). NaOH 3. OH OH (i) CHO NO2 (ii) OH Cl (iii) Br Cl CHAPTER 7 ANILINES 7.0 Introduction In chapter 6, phenols were discussed. It was noted that phenols are important intermediates in organic synthesis. In this chapter, anilines will be dealt with. These are compounds with an amino group attached directly to benzene. Most of its properties are similar to those of aliphatic amines including its smell. However presence of a benzene ring makes it different in some ways. Objectives By the end of this lesson, you should be able to: Explain the physical and chemical properties of anilines. Explain using equations different reactions shown by aniline. Describe how diazonium ion is prepared from various starting organic compounds. Describe reactions of diazonium ions. State some of the uses of anilines and their derivatives. 7.1 Preparation and properties of aniline When nitrobenzene is reduced by metal-acid reduction (Zn and HCl) aniline is formed. Reduction with sodium borohydride can also be carried out but in presence a catalyst poison Pd/C. They can also be prepared by heating azo compounds. Derivatized halobenzenes can also be a source of anilines formed through nucleophilic aromatic substitution reactions. NO2 NH2 Sn/HCl (Reduction) NO2 NH2 NaBH4 Pd/C Cl O2N O2N NO2 NH2 NO2 NH3 NO2 NO2 In certain situations, selective and special reagents may be required to bring about the reduction of the nitro group to amino group, depending on the type of group(s) attached to the benzene ring. A few examples are cited below. CH3 CH3 NO2 NH2 Sn/HCl CH3 NH4HS NO2 NO2 NO2 (NH3/HS) NO2 NH2 NH2 Ni/H2 200-4000C NHCOCH3 NHCOCH3 CHO Method used has when an the compound acid sensitive group CHO SnCl2/HCl NO2 NH2 Method used when the substituent has active unsaturation. Aniline has the following features: 1. Aniline is less basic than alkyl amines, due to resonance delocalisation of the lone pair of electron of nitrogen. 2. Basicity is enhanced by presence of electron donating group at ortho and para. Electron withdrawing groups make aniline less basic than aniline itself 3. Amino group (NH2) is a strong activator. Presence of alkyl groups attached to the ring reduces the activating power due to steric factors. 4. Electron withdrawing groups attached to nitrogen reduce the activating property. A good example is an amide group. The lone pair of nitrogen is not always available to activate the benzene ring, as it can also resonate with the double bond of the carbonyl. Nitrogen with a quaternary system (-NR3), like protonation of amino group, is a fully deactivating group. + NHCCH3 O NH=CCH3 O- 7.2 Reactions of aniline Aniline can undergo two types of reactions; those that involve the amino group and those that involve the benzene ring. 7.2.1 Reactions that involve the amino group NH-K+ K NH2 + NR3X- 3RX N CH+ 1. CHCl3 an isocyanide 2. 3OH- NH2 n-quaternary salt CHO N=CH + Schiff's base (Imine) NH2 N2+ NaNO2 H+/00C Diazonium ion NH2 (CH3CO)2O CH3COONa NHCOCH3 (N-Phenylethanamide) H2O/H+ NH-CH2 7.2.2 Reactions involving the benzene ring Aniline is too reactive for the halogenation reactions that polyhalogenation takes place at the para and two ortho positions. To introduce only one group, the amino group may be acetylated first. NH2 NH2 H2O + Br Br 3Br2 2,4,6-Tribromoaniline Br NHCOCH3 NH2 NHCOCH3 (CH3CO)2O NHCOCH3 Br2 Br + CH3COOH Br NH2 H2O 80% H+ 20% Br The ortho isomer can be prepared by introduction at the para position first. Meta substituted aniline product can be prepared from nitrobenzene. Iodo group to aniline occurs at para position due to selectivity principle due to steric factor. NH2 NH2 H2SO4 NHCOCH3 (CH3CO)2O HO3S HO3S Br2 H 2O NH2 NHCOCH3 H2O/H+ 100 0C Br HO3S NH2 I2 Br NH2 NaHCO3/H2O I NO2 NH2 NO2 Br2 Sn/HCl Lewis acid Br Br 7.3 Diazonium salts One of the most important reactions of anilines is the formation of diazonium salts. In turn diazonium ion can be converted to a variety of organic compound, which otherwise would require many steps to achieve from other aromatic compounds. N2+ Cl- NH2 NaNO2 + + NaCl H2O 2HCl / 00C Diazonium salt Diazonium salt undergoes two types of reactions; 1. Replacement of N2 (loss of N2) 2. Coupling reactions where N2 is retained 7.3.1 Replacement of N2 (loss of N2) Various nucleophiles can be used to replace the N2 group of the diazonium salt. Some replacements can be intermediate to other important aromatic compounds. Direct halogenation with fluorine is not possible but can be carried out from the diazonium salt reacted with fluoroboric acid (HBF4). Replacement of the N2 group can be carried out by use of hypophosphous acid (H3PO2) at low temperatures. N2+ :Z Z + Diazonium ion CuCl ArCl CuBr ArBr ArN2+ Diazonium ion CuCN KI ArCN ArI N2 Cl CH3 CuCl CO2H CN N2+Cl- NH2 CuCN CH3 HBF4 N2+BF4- CH3 H2O/H+ CH3 CH3 NaNO2 HCl / 00C F CH3 CH3 NaOH Heat OH CH3 H3PO2 H2O 0-250C CH3 + H3PO3 7.3.2 Coupling reactions where N2 is retained Diazonium salts undergo coupling reactions to form azo compounds. They are strongly colored compounds that have been used for a long time as dyestuffs. The aromatic compound that is undergoing attachment must have a powerful electron-releasing group like -OH, -NR2, -NHR and -NH2. N=N N(CH3)2 + +N2 SO3-Na+ SO3-Na+ (H3C)2N Methyl orange Red in acidic media Yellow in basic media OH + +N2 NO2 OH N=N 'Para Red' Dye NO2 7.4 Summary Aniline is less basic than alkyl amines due to resonance delocalisation of lone pair of electrons on nitrogen. Aniline shows reaction both at the amino group and at the benzene ring. Diazonium ion can be converted to many organic compounds which otherwise would require many steps. This makes diazonium ion very important in organic synthesis. 7.5 Questions and solutions Q1. By use of resonance structures and reaction mechanism, explain the following. (i) Introduction of an acetyl group (COCH3) to nitrogen reduces the activating properties of aniline. Q2. Suggest the major organic compounds (A-F) in the following transformations. N N Cl-+ OH (i) + A CH3 Cl2/H2O (ii) (iii) O2N B NH2 CH3 NH4HS (NH3/H2S) NO2 Q3. Outline synthesis of the following compounds. (i) (ii) (iii) 2,6-Dichlorophenol from phenol o-Nitroaniline from aniline m-chlorophenol from benzene Solutions 1. (a) See page 67 C 2. Cl OH N=N (i) Cl (ii) NH2 Cl CH3 H2N (iii) CH3 NO2 3. OH OH OH Conc. H2SO4 Cl2 (i) 1000C Cl FeCl3 (ii) (CH3CO)2O NHCOCH3 NHCOCH3 NO2 H2O/H2SO4 HNO3 H2SO4 H2SO4 0 100 C SO3H NO2 (iii) Cl2/FeCl3 HNO3 SO3H NO2 NH2 Sn/HCl H2SO4 OH- Cl Cl NaNO2/H+ OH N2+ClHCl/ 00C NaOH Cl Cl Cl 2). NaOH SO3H NHCOCH3 Cl 1). dil. H2SO4 Heat SO3H NH2 OH Cl NH2 NO2 CHAPTER 8 POLYNUCLEAR AROMATIC COMPOUNDS: NAPHTHALENE 8.0 Introduction In chapters six and seven, we looked at phenols and anilines and their derivatives and their importance as precursors for synthesis of many useful organic compounds. In this chapter will focus mainly on EAS reactions of naphthalenes, di-nuclear aromatic compounds and their derivatives. Orientation patterns of substituents on these compounds as well as their importance in organic synthesis will also be discussed. Objectives By the end of this lesson, you should be able to: Describe steps involved in synthesis of naphthalene from benzene Write down oxidation and reduction products of naphthalene Describe using mechanisms, EAS orientation patterns of naphthalenes Predict EAS products of naphthalenes with various nucleophiles State the effect on the rate of reaction and of a substituent on naphthalene on incoming nucleophile Discuss the reactions of substituted naphthalenes 8.1 Synthesis of Naphthalene Naphthalene has two benzene rings fused together. Naphthalene main source is coal tar. It has resonance energy of 61 Kcal mol-1. As has already been mentioned in chapter 2 that naphthalene has three carbon centers that are not equivalent. It is therefore expected to give more products during chemical reactions. This is a multiple steps synthetic route. It involves electrophilic substitution of benzene. This step works well if benzene is derivatized with an alkyl or halide groups. Second step involves ring closure while the third step is for aromatization of the extra-formed ring. O X + X AlCl3 O O Zn/Hg/HCl O X HO O X HO Clemmensen Reduction O Heat/Pd X O HF or H3PO4 N2H4/OH- X Heat Wolff-Krishner Reduction 8.2 Reduction and oxidation reactions of naphthalene Naphthalene can undergo oxidation and reduction reactions to give a variety of products depending on the reagent used. 8.2.1 Reduction Reactions When naphthalene is refluxed with ethanol in presence of sodium at 780C, the major product is 1,4-dihydronaphthalene. Addition of four hydrogens is achieved when it is refluxed with pentanol in presence of sodium at 1320C to form 1,2,3,4tetrahydronaphthalene. However, hydrogen in presence of metal catalyst nickel, platinum or palladium, naphthalene is fully reduced to decahydrodecalin. Na/CH3CH2OH Reflux (780C) Na/C5H11OH 0 Reflux (132 C) H2 1,4-Dihydro naphthalene 1,2,3,4-Tetrahydro naphthalene Decahydrodecalin Ni or Pt or Pd The mechanism of formation of 1,4-dihydronaphthalene follows the Birch reduction. The intermediate involved has the resulting negative charge and the radical are far apart and thus more stable. The alcohol supplies the hydrogens involved in the reaction. - Na. - . (-Na+) EtOH . More stable (-EtO-) . Na. (-Na+) EtOH (-EtO-) - 8.2.2 Oxidation of Naphthalene In presence of chromium trioxide at room temperature, naphthalene is oxidized to 1,4naphthoquinone. However, oxidation in presence of vanadium pentoxide (V2O5) at high temperatures, it is converted to phthalic anhydride. O CrO3/HOAc 1,4-Naphthoquinone (40%) 250C O O V2O5/O2 O 0 460-480 C Phthalic anhydride (76%) O 8.3 Orientation and EAS reactions of naphthalene Naphthalene is more reactive than benzene towards electrophilic aromatic substitution reactions. Electrophilic attack on naphthalene can occur at two positions, or . Orientation is controlled by the stability of the resonance structures involved as intermediates. If the aromaticity is destroyed in both rings, then that is unfavorable, and that route may not be followed. Normally the substitution is preferred over the substitution. It is noted that a attack leads to formation of more resonance structures where the aromaticity is lost in both rings unlike attack with more stable resonance arrangements. E E+ H + E E H H attack + Less stable + Stable Stable E H + Less stable + E E+ attack E + E H H Stable Less stable H + Less stable E H + Less stable 8.4 Electrophilic aromatic substitution reactions of naphthalene Most of the electrophilic aromatic substitution reactions of naphthalene are similar to those of benzene except for the fact that there are two different sites in naphthalene. Halogenation does not require the Lewis acid catalyst. Nitro group can be reduced to naphthalmine and in turn be converted to diazonium salt of naphthalene, which can lead to a variety of products as had been indicated. Bromonaphthalene, for example, can be converted to a Grignard reagent by reacting it with magnesium. This in turn can be converted to various compounds. Sulfonation of naphthalene, on the other hand, depends on the reaction temperature. At lower temperatures 1-naphthalene sulphonic acid, the kinetic product is formed. At high temperature, however, the major product is 2-naphthalene sulphonic acid, the thermodynamic product. Heating the kinetic product forms the thermodynamic product. NO2 HNO3 1-Nitronaphthalene (90-95%) H2SO4 Br Br2/CCl4 1-Bromonaphthalene (75%) Reflux Mg MgBr Alcohols Ketones acids etc Grignard Reagent SO3H Conc H2SO4 1-Naphthalene sulphonic acid (Kinetic product) 800C 1600C Conc H2SO4 SO3H 2-Naphthalene sulphonic acid (Thermodynatic product) 1600C In Friedel Crafts acylation the solvent used determines the products. When the solvent is tetrachloroethane (CH2Cl4), 1-acetonaphthalene is formed while use of nitrobenzene as the solvent leads to formation of 2-acetonaphthalene. COCH3 CH2Cl4 CH3COCl AlCl3 C6H5NO2 1-Acetonaphthalene (93%) COCH3 2-Acetonaphthalene (90%) The acetyl group can be converted to a carboxyl group by reacting the aceto product with bromine in a basic medium and later hydrolyzing in acidic medium. Alternatively, the process can be achieved by use of sodium oxochlorous (NaOCl) at 60-700C. COCH3 CO2H 1. Br2/OH+ 2. H+ CHBr3 1-Naphalene carboxylic acid COCH3 CO2H NaOCl + 60-700C CHCl3 2-Naphthalene carboxylic acid 8.5 Orientation of electrophilic substitution in naphthalene derivatives Introduction of a second substitution in naphthalene is more complicated than for benzene. The incoming group may be attached to any of the rings. However the following rules may assist in predicting where the substituent goes: a. An activating group on the ring tends to direct further substitution on the same ring. For example, the group is at position 1 () it direct the incoming group to position 4 more than position 2. If the group is at position 2 (), the incoming group is directed more to position 1. b. A deactivating group tends to direct further substitution on the other ring. If at position 1 () a nitro group will direct the electrophile to or of the other ring. Sulphonation depends on the reaction temperature. Some sample reactions are presented below. N2+Cl- OH OH NaOH + 4-Phenylazo-1-naphthol 0-100C N=N OH OH NO2 + HNO3 H2SO4 2,4-Dinitro-1-naphthol 200C NO2 N2+ClOH NaOH + 0-100C OH N=N 1-Phenylazo-2-naphthol Br CH3 CH3 Br2 in dark 1-Bromo-2-methyl naphthalene NO2 NO2 NO2 NO2 HNO3 + H2SO4/ 00C NO2 Minor NO2 Major NO2 H2SO4 SO3/ High temp HO3S 8.6 Summary Naphthalene can be reduced to various derivatives depending on the reagents and conditions used. Naphthalene is more reactive to EAS reactions than benzene and has two different positions ( and ). An activator on naphthalene directs the second substituent to the same ring while a deactivator directs it to the other ring. 8.7 Questions and Solutions Questions Q1. Suggest the major organic compounds (A-C) in the following transformations. OH Br2 (i) CH3COCl (ii) A B AlCl3 OCH3 NH2 H2SO4 (iii) 2000C/14 atm Q2. C Using a reaction scheme (step by step), show how the following conversions can be achieved. CO2H (i) Q3. By use of resonance structures and reaction mechanism, explain why 1Bromonaphthalene is exclusively formed and not 2-bromonaphthalene in monobromination of naphthalene Solutions 1. COCH3 Br (i) OH A= (ii) B= OCH3 OH (iii) C= 2. NH2 NO2 Sn/HCl HNO3 N2+ClNaNO2/HCl OH- 00C CuCN CN COOH H+/H2O 3. (a) See page 81-82 CHAPTER 9 ANTHRACENES AND PHENANTHRENES 9.0 Introduction and preparation In chapters 8 we looked at naphthalenes and their derivatives. In this chapter will focus mainly on anthracenes and phenanthrenes. They are tri-nuclear aromatic compounds and their derivatives. Orientation patterns of substituents on these compounds as well as their importance in organic synthesis will also be discussed. Objectives By the end of this lesson, you should be able to: Differentiate anthracenes and phenanthrenes in terms of structural features. Write down oxidation, reduction and Diels-Alder products of anthracene Describe EAS reactions of anthracenes and phenanthrenes. Describe positional activity of anthracenes and hence predict EAS products of anthracene with various nucleophiles. State some of the important derivatives of anthracenes. These are aromatic compounds with three rings fused together but differently. In anthracene the rings are linear while as in phenanthrene the fusion is tilted to one side. The resonance energy of anthracene is 84 Kcal mol-1 while that of phenanthrene is 92 Kcal mol-1. 8 9 1 9 2 7 3 6 5 10 1 2 7 4 6 Anthracene 10 8 5 4 3 Phenanthrene Anthracene can be prepared from phthalic anhydride and benzene in presence of Lewis acid. O O O + O AlCl3 O H2SO4 O OH O Phthalic anhydride Zn Heat Anthracene and phenanthrene undergo similar reactions. Most of their reactions involve the middle ring. 9.1 Oxidation and reduction reactions Oxidation of anthracene and phenanthrene with potassium dichromate in presence of an acid give 9,10-anthraquinone and 9,10-phenanthraquinone, respectively. Reduction involves the same position, where anthracene forms 9,10-dihydroanthracene while phenanthrene is reduced to 9,10-dihydrophenanthrene. O K2Cr2O7 9,10-anthraquinone Heat O K2Cr2O7 9,10-phenanthraquinone Heat O O Na/EtOH 9,10-Dihydroantracene Na/EtOH 9,10-Dihydrophenanthrene 9.2 Diels-Alder reaction Anthracene is able to undergo Diels-Alder reaction with maleic acid while phenanthrene does not. This reaction is used to differentiate the two compounds. O O O O O + O Maleic acid anhydride 9.3 Halogenation of anthracene Anthracene reacts with different chlorinating agents to give various chlorinated products. Chlorine gas in presence of sulphur carbide adds the two chlorine atoms with aromaticity in the middle ring being lost. However aromaticity is restored if the product is reacted with a base at high temperatures. Copper (I) chloride in presence of sulphur carbide introduces only one chlorine atom by substitution. On the other hand sulphonyl chloride (SO2Cl2) introduces two chlorine atoms by substitution. Cl OH- Cl2/CS2 Heat Cl Cl CuCl CS2 Cl Cl SO2Cl2 Cl 9.4 Sulphonation of anthracene Sulphonation is one set of reactions where the reaction involves either one or the two outer rings. Anthracene reacts with sulphuric acid in presence of glacial acetic acid to give two products at different proportions depending on the reaction temperature. At low temperatures 1-anthracenesulphonic acid is favored, while at high temperatures 2anthracene is formed at higher proportion. Excess sulphuric acid in absence of glacial acetic acid also yields two disubstituted products, where the amount of each depends on reaction temperature. Low temperature favors formation of 1,8-anthracene disulphonic acid while high temperatures favor the 2,7-analogue. SO3H SO3H H2SO4 + Glacial CH3CO2H 2- 1- Favored at high temperature Favored at low temperature SO3H SO3H Excess H2SO4 + SO3H HO3S 1,8- 2,7- Favored at low temperature Favored at high temperature 9.5 Alcohols of anthracene In anthracene, there are three different substitutable hydrogens. Therefore there are three different alcohols; 1-anthrol, 2-anthrol and 9-anthrol. These three alcohols have different physical properties as seen below. OH OH OH 1-anthrol 2-anthrol 9-anthrol Yellow solid Brown solid Yellow solid 0 mp 200 C 0 mp 152 C 0 mp 120 C 9.6 Positional activity of anthracene Electrophilic substitution reactions of anthracene, as has been mention earlier, can take place in three different positions, 1, 2 or 9. The stability of the resonance structures involved determines the product. From the resonance structures below, it can be deduced that substitution at position 2 least preferred. Position one ideally would be preferred over position 9 (or 10) because in 1 the intermediate resonance structures preserve naphthalene, which is more stable with 61 Kcal mol-1 as compared to 9 (or 10) where its benzene with 36 Kcal mol-1 is preserved. E E + E+ Position 9 or 10 + Aromaticity in 2 rings preserved E E+ E + Position 1 aromaticity preserved in 2 rings both resonance structures + E+ + + E E Position 2 Aromaticity of 2 rings destroyed 9.7 Anthraquinones These are most important derivatives of anthracene. They are colored compound that are used as dyestuffs. Many of them occur naturally are responsible of the coloration of some plant materials. In the laboratory, anthraquinone is prepared from phthalic anhydride and benzene in presence of a Lewis acid. However, commercially, anthraquinone is prepared from anthracene by oxidation with sodium dichromate in presence of sulphuric acid. O O O + H2SO4 AlCl3 CO2H O O (-H2O) O Phthalic anhydride O Anthraquinone (90-95%) Na2Cr2O7 H2SO4 Anthracene O The most interesting reactions involving anthraquinones are the reduction reactions. When it is treated with tin and hydrochloric acid in presence of acetic acid, anthrone is formed. When heated in presence of zinc and hydrochloric acid a bi-anthracene, biathryl is obtained. However, zinc in presence of a base leads to formation of 9,10-dihydro-9anthrol. O Sn/HCl CH3CO2H O Anthrone Zn/HCl Bianthryl O Zn/OH-/H2O 9,10-Dihydro9-anthrol OH 9.8 Summary Anthracene undergoes similar EAS reactions as naphthalene. Substituent at position 1 on anthracene is preferred over 2 and 9 because the intermediate resonance structures preserve naphthalene. Substitution at position 2 is least preferred. 9.9 Questions and solutions Questions Q1. Suggest the major organic compounds (A-C) in the following transformations. SO2Cl2 (i) A (ii) HNO3 B CH3CO2H (iii) 1). NaCrO7/H+ 2). Sn/HCl C CH3CO2H Q2 By use of resonance structures and reaction mechanism, explain why aromatic electrophilic substitution of anthracenes is preferred at position 9 over position 2. Solutions 1. Cl (i) A= (ii) B= NO2 Cl NO2 (iii) C= O 2. See pages 91-92 REFERENCES Adams, R., Johnsons; J., and Wilcox, C. (1968). Laboratory Experiments; 5th Edition, Macmillan Publing Company, New York. Brian, S. Furniss D. (1989). Vogels Text Book of Practical Organic Chemistry; 4th Edition. Bruice, P.Y. (1995). Organic chemistry; 1st Edition, Hall Inc, New Jersey. Clayden, Greeves, Warren and Wothers. (2001). Organic Chemistry; 1st Edition, Oxford University Press, New York. Fessenden, R.J and Fessenden, J.S. (1990). Organic Chemistry; 4th edition, Cole Publishing Company, Belmond. Finar, I.L (1973). Organic Chemistry; 6th edition. Longman, London. Hart, H., Craine, L.E. and Hart, D.J. (2003). Organic Chemistry –A short course. 11th Edition, Houghton Mifflin Company. Morrison, R.T and Boyd, R. N. (1987). Organic Chemistry; 5th edition, Longman, New York. Solomons,T.W.G. (1996). Organic Chemistry; 6th Edition, John Wily and Sons Inc, New York. Sykes, P. (1981). A guide to Mechanisms in Organic Chemistry. 5th Edition, Longman, London. PRACTICALS Electrophilic Aromatic Substitution Reactions Introduction Most electrophilic substitution reactions of aromatic compounds are thought to take place according to the following general mechanism:Using benzene as an example: H H + E+ Slow E E Fast + + H+ In some cases, the experimental evidence is interpreted best by proposing that E+, the electrophile is completely formed as a cation before it reacts with the aromatic compound as implied in the above equation. In other cases, the data are best interpreted in terms of simultaneous attack of aromatic system on E-X and loss of the leaving group H H E E Slow + E-X + + X- Fast + H+ For convenience however, electrophilic aromatic substitution reactions are often interpreted in terms of attack by previously formed E+, with the mental reservation that this may not always be the best representation of reaction The experiments that follow have been selected to illustrate some of the important aspects of electrophilic aromatic substitution reactions (EAS) EXPERIMENT 1: PREPARATION OF NITROBENZENE Introduction The conditions used in the nitration of an aromatic compound depend on the structure of the compound. The nitrating agent in most cases appear to be nitronium ion:O N O+ The source of nitronium ion may be a nitronium salt, such as nitronium fluoroborateNO2+BF4- or as in this experiment, the ion may be derived from nitric acid by reaction with conc. sulphuric acid. i) ii) HNO3 + H2SO4 +NO2 + C6H6 Overall reaction: C6H6 + HNO3 +NO2 + H2O + C6H5NO2 + HSO4H+ C6H5NO2 + H2O Nitrobenzene Procedure Before you do the experiment, note the following: The benzene and acid layers are only slightly soluble in one another. When benzene dissolves in the acid layer, it reacts and swirling speeds the process of solution. Nitrobenzene is appreciably toxic and its vapour should not be allowed to escape in the atmosphere (keep the experiment in the fume hood). The liquid is also toxic by skin absorption and should be washed if spilled on the body with methylated spirit followed by soap and warm water. Benzene is toxic: Avoid excessive contact with either liquid or vapour, do the experiment in the fume hood. Place 8ml of conc. Nitric acid in 50 ml flask and add slowly 9 ml of conc. Sulphuric acid, swirling gently and cooling the flask under running tap water. Then introduce, 7 ml (0.075 mole) of benzene in small portions from a measuring cylinder. By constant swirling and brief cooling periods in cold water, keep the temperature of the mixture close to but not exceeding 60oC and not below 55oC, during the addition of benzene. When all the benzene has been added, attach a reflux condenser and heat the mixture on a water bath at 60oC for 30 minutes. From the time to time during this period lift the flask and condenser out of the water bath, and swirl the reaction mixture vigorously to ensure good mixing of the immiscible layers. At the end of refluxing, cool the flask in cold water. Pour the complete reaction mixture into about 60 ml of cold water in a beaker (200 ml), and swirl the mixture well order to wash out as much acid as possible from nitrobenzene and allow to stand. When nitrobenzene has settled to the bottom, pour off the acid liquors completely as possible and transfer the residual liquid to a separatory funnel. Add 10 ml of 1.5 M sodium carbonate solution, shake gently, frequently releasing the pressure. Allow to stand and then remove the upper aqueous layer. Repeat the process with two further 10 ml portions of sodium carbonate solution, followed once by 10 ml portion of water. Remove the upper aqueous layer in each case and retain the lower Transfer the nitrobenzene to a stoppered test-tube or small conical flask and add a few small lumps of anhydrous calcium chloride, swirling occasionally until the solution is clear. Set up the apparatus for distillation using air condenser and distil the nitrobenzene, collecting the fraction boiling at 198-202oC. Do not distil to dryness nor allow the temperature to rise above 205oC, for there may be a residue of m-dinitrobenzene and higher nitro compounds and an explosion may occur. Record your yield and determine the refractive index of the nitrobenzene Questions 1. Outline, using equations production an electrophile using conc. Nitric and sulphuric acids. 2. Using appropriate structural formula, indicate the mechanism of nitration of benzene. 3. Nitration by nitric acid alone is believed to proceed by essentially the same mechanism as nitration in the presence of sulphuric acid. Write the equation for the generation of nitronium ion from nitric acid alone. 4. Explain why nitrations are usually carried out at comparatively low temperatures. EXPERIMENT 2: Reduction of nitrobenzene to Aniline Introduction Aromatic nitro compounds may be reduced to the corresponding aromatic amines by many reagents. In this experiment, nitrobenzene is reduced to aniline by nascent hydrogen, which is generated from tin and conc. hydrochloric acid. C6H5NO2 + 6 H C6H5NH2 + 2H2O This reaction is of particular importance as a step in the conversion of benzene to a very large range of useful compounds that can be prepared from aniline. Procedure [Note: Ensure each joint is smeared with small amount of lubricant preferably silicone grease in order to prevent seizure of the joints when using strongly alkaline solution] Place 3 ml (0.03 mole) of nitrobenzene and 7 ml of granulated tin (small pieces) in 100 ml conical flask. Measure 15 ml of conc. Hydrochloric acid. Pour about 4 ml of the acid down the condenser, and steadily swirl the contents of the flask. The mixture becomes warm and before long the reaction should be quite vigorous; if it boils vigorously, moderate the reduction somewhat by momentarily immersing the flask in cold water. When the initial reaction slackens on its own, add another few mls of acid; continue to swirl the flask, cool again if the reaction becomes too violent. Do not cool more than is necessary to keep the reaction under control; keep the reaction mixture well mixed. Proceed in this way until all the acid has been added. When all the acid has been added and the vapour of the reaction has subsided, heat the flask in a boiling water bath for about 30 minutes. This completes the reduction of nitrobenzene to aniline by stannous chloride. If after 30 minute heating, some nitrobenzene still remains, as is evidenced by oily droplets on the surface of the reaction mixture and by the characteristic smell of nitro compounds, add a few more of small pieces of tin and 2-3 ml of conc. Hydrochloric acid and continue heating on the water bath until a homogenous solution is obtained. The aniline is now present as complex salt, aniline chlorostannate, which although appreciably water- soluble may separate during the reduction, particularly during the cooling which follows. Cool the reaction mixture to room temperature. In a 100 ml conical flask, make up a solution of sodium hydroxide (11.5 g in 15 ml water) and cool it to room temperature also. Add the sodium hydroxide gradually and with gentle swirling to the cooled reaction mixture. If the resulting mixture boils during the addition of the alkali, cool again. Keep the reaction mixture in ice-cold water for about 5 minutes. The aniline, which is now present as the free base, will now be visible as a distinct upper layer. Decant off, from any residue solid, both the aqueous and aniline layer in 50 ml quick fit flask. Equip the flask for steam distillation using Claisen still head. Steam distil by heating the flask directly until no more drops of aniline come over. Collect the distillate in a separating funnel Add 3 g of sodium chloride to the separating and shake. This reduces the solubility of aniline in water (about 3%). Run off the crude aniline into a test-tube. Add a few pellets of sodium hydroxide to remove moisture, and allow to stand in a stoppered tube until turbidity disappears. Determine the refractive index of your aniline, a confirmation of its identity. Questions 1. Outline with the aid of appropriate chemical equations, the various stages in the reduction of nitrobenzene to aniline using tin and hydrochloric acid. 2. Give two alternative reagents that can be used to reduce nitrobenzene to aniline other than metal-acid reagents. 3. Explain why calcium chloride cannot be used as a drying agent for aniline. 4. State the reagents you would use to convert 1-methyl-2,4-dinitrobenzene into the following; a) b) 2-amino-1-methyl-4-nitrobenzene. 1-amino-4-methyl-2-nitrobenzene. EXPERIMENT 3: Nitration of Aniline to p-Nitroaniline Introduction The amino group activates benzene ring towards electrophilic reagents to a greater extent than the hydroxyl group, and aniline is very readily oxidized by nitric acid, so direct nitration is impossible. It is therefore necessary to protect the amino group while carrying out the nitration. This is conveniently done by acetylation. The acetyl group does not change the position, to which the substituent is directed, but it is fairly a large group and hence, the yield is nearly all p-nitroacetanilide. The acetyl group is finally removed by alkali hydrolysis according to the following equation NH2 NHCOCH3 (CH3CO)2O HNO3 CH3CO2H H2SO4 NHCOCH3 NHCOCH3 NaOH NO2 NO2 Procedure Step 1: Preparation of acetanilide Put 5 ml (0.05 mole) of aniline, 7 ml ethanoic anhydride (in excess) and 5 ml of concentrated ethanoic acid into a 50 ml quick fit flask fitted with water condenser. Reflux the mixture for 15 minutes. Cool the mixture and then pour it with stirring into 50 ml of water in a 250 ml beaker. The acetanilide will solidify into white lumps. Cool the mixture, filter off the solid at the pump, wash it with water and finally dry it in the oven at 90-100oC (mpt of acetanilide is 114oC) Step 2: Nitration of Acetanilide Put 2.5 ml of conc. Ethanoic acid into a 100 ml conical flask and add with stirring 2.7 g (0.02 mole) of acetanilide followed by 5 ml of conc. Sulphuric acid. Cool the hot clear solution in a freezing mixture of ice and sodium chloride, stirring gently with a thermometer until the temperature is between 0 and 5oC. Now, add drop wise with stirring, 1.5 ml of fuming nitric acid, keeping the temperature below 25oC When all the acid has been added and temperature begins to fall, remove the solution from the freezing mixture and allow to stand for ten minutes. If the temperature starts to rise steadily, return the solution to the freezing mixture for a while. Now pour the solution into 50 ml of water contained in 250 ml conical flask. The pnitroacetanilide will precipitate as a yellow crystalline powder. Filter off at the pump, wash with water and drain thoroughly Step 3: Hydrolysis of p-nitroacetanilide Transfer the crystals of p-nitroacetanilide to a 50 ml quickfit, add 8 ml of ethanol and a solution of 1 g of NaOH in 5 ml of water. Reflux the mixture for about ten minutes, after which there should be a clear yellow solution. Cool the flask to crystallize the p-nitroaniline Filter at the pump, using the filtrate to wash out the last part of the solid from the flask, and wash with a little water. Drain thoroughly, recrystallise from the minimum quantity of aqueous Ethanol and dry the product. Determine the melting point of the pure pnitroaniline and calculate the molar yield based on the amount of acetanilide used. Questions 1. Give an alternative reagent to the one you used in the above practical; for protecting the amino-group during nitration. 2. If direct nitration of aniline is carried out in strongly acidic medium, a considerable % of the amine is, as expected converted to a complex variety of oxidation products. Such nitration as does occur however, results in mnitroaniline as the major product rather than para-ortho isomers that might have been expected due to the presence of NH2 group. Explain. 3. Explain why -NH2 group activates the ring more than the -OH group. 4. Outline, giving reagents employed, how you would convert p-toluidine(4methylaniline) to 4-amino-3-bromotoluene. 5. Arrange the following functional groups in increasing order of their activation of aromatic rings towards electrophilic reagents: -NHCOCH3, -NH2 -NHCH3, -OH, -OCH3, -SH, -SCH3. EXPERIMENT 4: Preparation of Iodobenzene Introduction It is difficult to iodate benzene because the reaction between benzene and iodine is reversible:C6H5NO2 + I2 C6H5I + HI The most satisfactory way of attaching a halogen atom to an aromatic ring is by replacement of the diazonium group, which is in turn obtained from the corresponding primary amine. Replacement of diazonium group by an iodine atom is readily effected by direct reaction of iodine with the appropriate diazonium compound. Thus iodobenzene is usually prepared by the reaction of benzene of diazonium chloride and potassium iodide. [C6H5N2 ]+Cl- + KI C6H5I + KCl Iodobenzene is purified from involatile inorganic impurities by steam distillation, as iodobenzene is almost insoluble in water. To obtain a good separation, the distillate is shaken with several portions of diethyl ether, as the partition coefficient for iodobenzene favors the either layer. Final purification is by distillation. Procedure Step 1: Preparation of benzene diazonium chloride solution Place 4 ml (0.4 mole) of aniline in a 100 ml flask, and add a mixture of 12 ml of conc. HCl and 12 ml of water. Swirl gently to dissolve the aniline. Place a thermometer in the flask and cool the mixture in an ice-salt mixture. Dissolve 3.4 g of sodium nitrite (NaNO2) in 4 ml of water in a test-tube, and cool in the freezing mixture. Introduce the mixture slowly into the flask. Stir the mixture with the thermometer and do not allow the temperature to rise above 10oC. The flask now contains a solution of benzene diazonium chloride. Step 2: Preparation of iodobenzene [Note: Ether is very flammable] Dissolve10 g of potassium iodide in 12 ml of water in a test-tube and cool in the freezing mixture. Add the solution to the benzene diazonium chloride solution, keeping the flask in the freezing mixture not to allow the temperature to rise above 10oC. When all the KI solution has been added, transfer the flask to a cold water-bath and fit a reflux condenser After ten minutes, bring the bath to a boil and maintain it at this temperature for about 15 minutes. Nitrogen is evolved and dark oily drops of impure iodobenzene separates out. Steam distil the mixture until no more oily drops appear in the distillate. Extract the iodobenzene from the distillate with three successive 5 ml portions of diethyl ether, using a separating funnel.. Combine the ethereal extracts and wash them with 5 ml of 10% NaOH and then with 5 ml of water. In each case reject the washings. Dry the ethereal solution of iodobenzene by allowing it to stand in contact with little fused calcium chloride in a stoppered test-tube until the solution is clear. Distil off the ether from hot water bath. [Extinguish all flames in the lab]. Remove the hot water-bath and distil the iodobenzene using the condenser as an air condenser. Collect the fraction distilling between 178oC and 184oC. Determine the refractive index of iodobenzene and calculate the molar yield based on the aniline used. Questions 1. Outline the synthesis from benzene diazonium chloride, the following compounds:a) Chlorobenzene b) Flourobenzene d) Benzoic acid e) Benzene 2. Outline the synthesis of m-nitrobenzene from toluene. c) Phenol EXPERIMENT 5: Preparation of methyl orange (p-(p-dimethylaminophenylazo) benzene sulphonic acid, sodium salt) Introduction Under the correct conditions, diazonium salts react with certain aromatic compounds to yield products of the general formula Ar-N=N-Ar, called azo compounds. In this reaction known as azo- coupling, the nitrogen of the diazo group is retained in the product, in contrast to the majority of reactions involving diazonium salts ArN2+ +Ar’H Ar-N=N-Ar’ Azo compound + H+ The aromatic ring (Ar’H) undergoing attack by diazonium ion must, in general contain a powerful electron releasing group because diazonium ion ArN2+ is only weakly electrophilic and thus only capable of attacking very reactive rings. Coupling is a further example of EAS, in which the substitution usually occurs para to the activating group (R). R R + ArN2+ R + H+ + H N=NAr N=NAr Azo compounds are strongly colored: are yellow, orange, red, blue or green depending on the exact structure of the molecule. Hence their great importance as dyes. Color changes also occur depending on the pH, thus their use as acid base indicators, for example methyl orange. Procedure Step 1: Diazotisation of sulphanilic acid In a 250 ml conical flask, dissolve by boiling 4.8 g of sulphanilic acid crystals in 50 ml of 2.5% sodium carbonate solution. Cool the solution under the tap, add 1.9 g of sodium nitrite and stir until it is dissolved. Pour the solution into the flask containing about 25 g of ice and 5 ml of conc. HCl. In a minute or two, a powdery white precipitate of diazonium salt should separate and is to be used in this form as its stable for a few hours. Step 2: Preparation of methyl orange In a test-tube, thoroughly mix 3.2 ml (0.025 mole) of dimethylaniline and 2.5 ml of glacial ethanoic acid. To the suspension of diazotised sulphanilic acid contained in a 400 ml beaker add, with stirring the solution of dimethylaniline acetate. Wash out the test tube with some of the diazotised sulphanilic acid solution to ensure complete transfer of dimethylaniline acetate. Stir and mix well and within a few minutes, to red acid stable form of the dye should separate.. a stiff paste should result in 5-10 minutes and 35 ml of 10% NaOH is then added to produce the orange salt. Stir well and heat the mixture to boiling point, when a large part of the dye should dissolve. Place the beaker in a pan of ice and water and let the solution cool undisturbed. Collect the product on a buchner funnel using saturated NaCl rather than water to rinse the flask and to wash the dark mother liquor from the filter cake. Recrystallise the crude product from water, after making preliminary tests to determine the optimum conditions. Calculate the molar yield based on the dimethylaniline used. Questions 1. Write equations for: a) Diazotisation of sulphanilic acid b) Conversion of diazotised sulphanilic acid to methyl orange 2. The efficiency of the coupling reaction between diazonium salts and other compounds is very dependent upon reaction conditions and particular the pH. Explain why this should be so. 3. An azo compound is readily cleaved at the azo linkage by tin (II) chloride to form two amines. What is the structure of the azo compound that is cleaved to 4-amino-3bromotoluene and 4-amino-2-methylphenol? EXPERIMENT 6: Preparation of p- Di-t-butyl benzene Introduction Introduction of alkyl groups into an aromatic ring is a useful process and is carried out on an industrial scale to produce compounds such as cumene (1-mthylethylbenzene) Alkylation involves attack of an electrophilic carbon species on the ring, choice of the reagent and conditions depend on the alky group and the reactivity of the ring. In this experiment, the major product of the reaction between benzene and t-butylchloride is p-dibutylbenzene C(CH3)3 + 2 (CH3)3CCl AlCl3 C(CH3)3 Procedure In a thoroughly dry 100 ml conical flask, place 10 ml of t-butylchloride and 5 ml (0.06 mole) of benzene. Arrange a trap for HCl gas by connecting a length of rubber tubing from a one-hole rubber in the flask to a glass funnel inverted just below the surface of a beaker containing water Obtain 0.5 g of anhydrous aluminium chloride in a stoppered test tube. Minimize exposure of this compound to air, the proper amount can be estimated by comparison with weighed demonstration sample. Cool the benzene t-butyl chloride mixture in an ice bath (clamp the flask loosely to avoid tipping). Add about one-third of the aluminium chloride, and swirl the flask in ice bath. After bubbling has occurred for 4-5 minutes add the rest of aluminium chloride in two separate portions during the next 10-15 minutes. (restopper the flask as quickly as possible and rinse your fingers, since brief exposure to the acid fumes cannot be avoided) When the reaction begins to subside, remove the ice bath and allow the mixture to warm to room temperature. Unstopper the flask and add 10 ml of ice-cold water. Then add about 10 ml of diethylether (extinguish all the flames), swirl the mixture and transfer the contents of the flask to a separating funnel; rinse the flask with small amount of ether and add the washing to the separating funnel. Gently swirl the funnel for a few minutes taking care to release the pressure frequent intervals. Separate the ether layer, wash it with two 5 ml portions of water ad dry it with a small amount of anhydrous magnesium sulphate in a stoppered 50 ml quick fit flask. Distil off the ether from hot water bath (put off all the fire in the lab). Using the same flask, but with a reflux condenser added recrystallise the p-di-butylbenzene from 10 ml of methanol, collect and air dry the product, record its yield and its melting point. Calculate the molar yield based on the benzene used. Questions. 1. State briefly any four limitations to the use of Friedel-Crafts alkylation. 2. GLC analysis of the crude reaction mixture from the experiment shows in addition to unreacted benzene and p-di-butylbenzene, three other products. Suggest structures for these by products and write equations for their formation. 3. Other than the comparison of melting points, how would you, using chemical method, prove that the product you isolated in the experiment was p-di-butylbenzene?