PLASTICITY OF GLASSY POLYMERS: NEW EXPERIMENTAL
advertisement

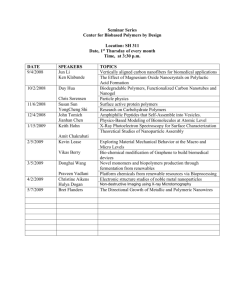
PLASTICITY OF GLASSY POLYMERS: NEW EXPERIMENTAL FACTS DISREGARDED BY CURRENT THEORIES AND COMPUTER MODELLING E.F.Oleinik, Institute of Chemical Physics Russian Academy of Sciences, MOSCOW, RUSSIA Today there are several theories of the molecular level plasticity for polymeric glasses and quite a lot of computer simulations analyzing the processes. All these are surprisingly uniform in the main point of interest: they all analyze an orientation and uncoiling of chains through conformational rearrangements, redistribution of different conformers along the chain, local and collective segmental dynamics, formation of kink-type structures in chains and some other intrachain processes as main factors controlling plasticity of polymer glasses in the bulk. The approach is helpful if the reorganization of chains is really the main factor controlling plasticity. However, if it is not the case? Than the current theories and computer modeling results play with the “secondary importance” structural transformations appearing in glassy solids during deformation. Rate controlling step of plasticity will be not clarified and understood in a such way. In this presentation the experimental data manifesting that intra-chain structural transformations do not control the plastic response of solid polymers, at least for glasses built from flexible and semiflexible chains, will be presented. Measurements of the energy of cold work for glassy polymers permitted us to suggest that the key rearrangements have an intermolecular origin. Nucleation of local shear transformations (ST) trough the displacements of the short molecular segments of neighbor chains is the event controlling a development of the following plastic process in glassy matter. Conformational rearrangements in chains appear only after an ST formation and proceeds as a local relaxation transformation providing the release of an energy excess accumulated by this ST. Such second process is going easy and does not control the plasticity kinetics. Facts supporting this view will be presented. All solids, crystalline and glassy, store the energy of cold work. For glassy polymers it was found that at deformation stages below the y (the macroscopic yield strain) 100% of Wdef expended for sample deformation is transformed into a sample internal energy Udef. There is no deformation heat Qdef at these strains. This stored energy appears in a sample not due to appearance of new conformers in chains. Stresses and strains are yet too small for such conformational rearrangements. It looks that the nucleation of the plasticity carriers is the key process (like nucleation of dislocations in crystals) at this loading stage. The formation of such carriers is the most difficult step in whole process. And namely STs are the main carriers of plasticity in glass. There is other interesting feature in plastic behavior of glassy polymers. Well plastically deformed glass contains two different inelastic strain components. One component is carrying out nearly all Udef. In the thermally stimulated residual strainrecovery experiments this component disappears from a sample up to 200-3000C and even more below Tg (low temperature recovery, LTR, component), i.e. in the glassy state of a deformed polymer. It is quite difficult to imagine that new conformers (appeared at straining) in chains able to recover in a glassy state at such low temperatures. And the other component is recovered for all polymers in Tg temperature interval (high temperature recovery, HTR, component). Appearance the HTR component manifests the structural recovery of chains enriched by the extended conformers, which had appeared during a sample loading. It looks natural that the conformational transformations happen in deformed chains at Tg interval. It appears that the carrying STs strain component in the deformed glass (LTR component) is necessary pre-cursor for appearance of any conformational rearrangements in chains. It was shown in the following way. If one will erase only the LTR component from a deformed glassy sample by its heating from Tdef to a little below Tg and will reload the sample again at the same Tdef, new additional strains in a sample will appear at first only in the form of LTR, ST containing strain component, non conformational one. Intensity of the HTR component is not growing at the beginning of such reloading. Only when the LTR component at the reloading will reach the critical level of intensity, the conformational strains (intensity of the HTR peak) starts to grow. The result shows that the conformational rearrangements in polymer glass during its loading never happens without formation of their pre-cursor, the STs. There are several other interesting features of the deformation energy storage processes in glassy polymers. For example, the fraction of the energy stored by a deformed sample is decreasing with total strain εdef. The same exists in crystalline metals. What are the processes decreasing the concentration of deformation defects at high εdef? And what is the nature of deformation heat, Qdef, in the plasticity of polymer glasses? It can’t be the same origin as in crystals. In our view of the plasticity process in glasses Qdef is the result of the release of energy accumulated by STs. However, such release appears only together with local conformational rearrangements in chains surrounding given ST. New theories and simulations should not disregard all described and other energy storage experiments in glassy polymers plasticity. It looks that only trough modeling and analyzing such processes one can clarify the nature of structural rearrangements, their scale and mechanisms of plastic deformation in glassy polymers. At the end of the presentation simple MD modeling the shear deformation of the 2D atomic Lennard-Jones glass will be shown. The modeling demonstrates the sequence of elementary plastic events appearing during shearing of the computer glass; nucleation of local STs, their coalescence and formation of macroscopic (sample scale) shear band. The work is supported by the Russian Foundation for Basic Researches (Grant № 05-03-32481) and by the Program № 3 of the Chemistry Division of the Russian Academy of Science, 2005.