All about HCL
advertisement

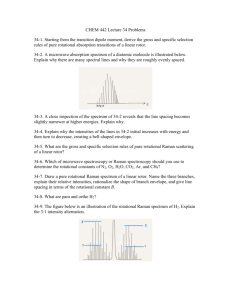
Chem 327 The Rigid Rotor and HCl Rovibrational Transitions Renember , throughout this whole discussion, the symbol L for angular momentum and J for angular momentum mean exactly the same thing- as to the quantum numbers associated with them. By convention, we use the letter J to indicate that we are looking at rigid rotor problems and L to indicate that we are looking at atoms, but I don't make that distinction here- I treat them interchangeably. Problem We’ve talked in class about the particle on a ring as a model of rotation in 2 dimensions. In that case, we had only to worry about the component of angular momentum along a particular axis, usually taken to be the z axis, because this angular momentum Jz (or lz) can only point in the +z or the –z direction. Although there are some physical situations that are similar to the particle on a ring problem, more common are examples of the usefulness of 3D particle on a ball problem otherwise known as the rigid rotor. Recall that like the particle on a ring, the quantization of angular momentum and thus of rotational kinetic energy is not due to the imposition of some external potential, V, as it is in for example the harmonic oscillator, but rather is a result of the fact that the particle is confined to a ‘sphere.’ Note: remember that what we are almost always talking about when we discuss the rigid rotor is two bodies moving relative to one another each at a fixed distance (the rigid part of the rigid rotor) from their mutual center of mass. Through the use of reduced mass and center of mass coordinates, we can mathematically transform this situation into something that looks like a particle confined to a sphere of constant radius r where r is the distance between the atoms. Such is similar to what we have already done for the harmonic oscillator and particle on a ring. In a previous problem set for the particle on a ring, we saw that the form for the angular momentum operator for that system was particularly simple, as was the form of the wavefunction (look at the problem set). Unfortunately, this simplicity is lacking in our analogous 3D problem. Instead of just one angular variable in the case of the particle on a ring, , the wavefunction for the rigid rotor is a function of two variables, and . We have talked about the meaning of these variables before and they are defined in your textbook, suffice it to say here that as in the P.O.R. is defined as the angle around the z axis lying in the xy plane while is the angle from the z axis toward the xy plane. The solutions of the Schrodinger equation for the particle on a ball are eigenfunction of the total angular momentum squared J2 and of the component of angular momentum along the z axis, Jz. Because of the former, they are also eigenfunctions of the rotational kinetic operator which looks like: J 2 2 2I 2I 1 1 2 Sin Sin 2 Sin Where J is the angular momentum operator and I=r2 is the moment of inertia of the system. Don’t worry, you’ll never have to use this operator. Do take note, however, of the fact that the part of this operator that depends on is very similar to that in the particle on a ring problem, it’s just got a lot of that other crap that depends on with it. In fact, the operator for total angular momentum about the z axis in the rigid rotor is exactly the same as it was for the particle on a ring (look it up). Any these solutions are called the spherical harmonics, Y with quantum number m and l. Yl m , NPl m Cos eim part that depends on only, looks exactly like particle on ring eigenfunctions normalization constant associated Legendre polynomial (this is polynomial in the variable Cos Table 1. The first few assoicated Legendre Polynomials Associated Legendre Polynomial Pl m Cos P00 Cos 1 P10 Cos Cos P11Cos 1 Cos P20 Cos 3Cos 1 P21 Cos Cos 1 Cos2 2 Cos Sin P22 Cos 1 Cos Sin 2 1 2 Sin 2 1 2 2 The eigenvalues of L2 for the rigid rotor are given by l l 1 2 and for rotational angular momentum they are 2 l l 1 2I for quantum number l (=0,1,2,…). Eigenvalues of Lz are ml for ml=-l,-l+1,…,l. Note that these values imply that every rotational eigenfunction of quantum number l is 2l+1-fold degenerate. These 2l+1 degenerate functions differ only with respect to the orientation of their angular momentum component along the z axis. Please see Figure 1 for details. z ml=+ ml=0 ml=- y L x L2 l l 1 2 L ll 1 Figure 1: An illustration of the vector model for angular momentum in a rigid rotor ( or an atom for that matter) for the state with quantum number l=1. Here, angular momentum is given the label L instead of J, they are frequently used to mean the same thing So, the rotational motion of molecules can be treated as rigid rotor problems (particles on a ball). In general, rotational transitions of molecules are relatively low energy processes and occur in the microwave region of the electromagnetic spectrum. One thing to keep in mind though is that not all molecules that rotate (which is all of them except ‘monatomic’ molecules like He) will give rise to a microwave spectrum. So see why this is the case, recall a similar situation for the harmonic oscillator. We talked then about how although all molecules vibrate, they have some zero point energy because of the uncertainty principle, not all molecules have spectra that show up in the infrared. This is because we required that for a molecule that vibrates, the vibration itself must give rise to a changing dipole moment in order to interact with the electromagnetic field and thereby absorb the IR radiation. Such is also the case rotating molecules, except here the bond itself must have some permanent dipole so that rotation of that dipole looks like an oscillating dipole to photons that happen by. Problem 1 Derive an expression for the difference in rotational kinetic energy between consecutive states, i.e. between the states with quantum number J and J+1. This should end up being a simple expression proportional to J. What follows is an example of the analysis of a molecule that both vibrates and rotates at the same time. You’ll see that we can get a great deal of information about the system using these two different spectroscopic techniques. All about HCL Figure 1. The rotational/vibrational spectrum of HCl Explanation of Figure 2 This spectrum results from absorbtion of radiation by simultaneously vibrating and rotating HCl molecules and some features of it bear description. First, you should understand that all of the lines in this spectrum involve the excitation of HCl from the ground vibrational state (q.n. v=0) to the first excited state (q.n. v=0). The fact that there are multiple lines that range from about 7.8 x1013 s-1 to 9.20 x 1013 s-1 simply is a reflection of the "width" of the vibrtional peak. You are not accustomed to seeing a wide vibrational feature, for instance in a IR spectrum, that is broken up into separate lines. Rather you are probably more used to seeing a single broad feature. This spectrum of HCl was taken in the gas phase, most of the IR spectra you see are taken in the liquid or solid phases which has the effect of blurring the lines together. The fact that there are multiple lines within this vibrational band results from the fact that when the vibrational transition occurs, a change in the rotational kinetic energy (angular momentum) occurs at the same time. You'll also note that there are essentially two halves to the spectrum, the set of lines on the left and the set of lines on the right which are separated by a rather larger gap centered at about 8.65 x 1013 Hz. Aside from the large gap, the spacing between the lines on both the right and left halves are is approximately equal (about 0.070 x 1013 s-1). The lines on the right come at a somewhat higher energy and involve the molecule going from the ground vibrational state and a lower rotational state (for example, v=0 and say J=0) to the first excited vibrational state and a higher rotational state (v=1 and say J=1). On the other hand, the set of lines on the left correspond to an excitation from the lower vibrational state and a higher rotational state (eg v=0 and say J=1) to the excited vibrtational state and a lower rotational state (v=1 and say J=0) Therefore, you can interpret the center of the spectrum (at 8.65 x 1013 s-1) to correspond to the vibrational excitation from v=0 to v=1 that occurs without any changes in the rotational kinetic energy or quantum number J, i.e J=0. Notice that there is no peak corresponding to such a pure vibrational transition not accompanied by a change in J. In other words, for the transition to show up in the spectrum J must change, and in fact it must change by exactly 1 unit J=1. So, peaks in the spectrum that correspond to energies slightly higher than the missing pure vibrational transitions are caused by the vibrational excitation occurring in conjunction with an increase in J (J=+1) while those peaks that correspond to energies slightly lower than the pure vibrational transition correspond to the case where the rotational kinetic is lower in the higher vibrational state than in the ground vibrational state (J=-1). This is an example of what is known as a selection rule and these arise because of the detailed way in which the electromagnetic radiation interacts with the rotational or vibrational wavefunction. Short elaboration: Photons causing vibrational transitions that also involve an increase in the rotational kinetic energy must not only have the energy required to vibrationally excite the molecule but have to have an additional (smaller) bit of energy to increase the rotational energy as well. On the other hand, if the vibrational transition involves going to a lower rotational state, then some of that extra rotational energy in the ground state can be used to help the vibrational transition and therefore the energy of the absorbed photon can be that much smaller. . Question Finally, you’ll see that each of the peaks in Figure 2 is slightly split. Can you come up with a plausible explanation for why this is so? Note, I didn't state what isotope of Cl I was using to get this spectrum. Problem 2 We've talked a little bit about the vibrational spectra of molecules under the harmonic oscillator approximation, which ignores the effects of 'anharmonicity' discussed. Using the fact that the center of the ‘absent’ peak of the spectrum in Figure 2 corresponds to the unallowed pure vibrational transition to find the fundamental frequency, 0, force constant k, of the HCl vibration. Also find the zero-point energy of HCl in its ground state. Problem 3 Use your result from the Problem 1 in this set to derive an expression relating the spacing between rotational energy levels (~0.070 s-1) and the value of I, the moment of inertia. In problem 1, you derived an expression for the difference in energy between successive rotational energy states. These energy differences, of course, correspond to the energies of the photons required to effect the rotational transitions. Now, take note of the fact that the spacing between the lines in Figure 2 corresponds to the differences between the energies of the photons. Derive an expression that relates this spacing between these rotational lines (which amounts to about 0.070 x 1013 s-1) to differences in the energies of the photons and ultimately to the value of the moment of inertia I for the molecule. Once you have I, you should be able to find the H-Cl bond length, r. Question Okay, so the spacing between rotational energy levels in Figure 2 is not exactly equal throughout. Can you think of why this might be so?