N-Heterocyclic Carbenes - Denk Group
advertisement

N-Heterocyclic Carbenes1
Stable N-heterocyclic carbenes are important areas of study. Understanding the structure
and reactivity of these highly Lewis basic (one of the strongest known neutral bases) is of much
interest, especially since they are expected to be alternatives and extensions to more basic
phosphanes. Stable ylidene carbenes are also used to prepare main group and transition metal
complexes.
N-heterocyclic carbene complexes show promising properties for a number of catalytic
reactions in organic chemistry, especially since imidazolium salts are cheap to synthesize for
industrial applications. N-heterocyclic carbenes have been overlooked even though they are
catalytically relevant ligands in coordination chemistry, and will play a major role in future
organometallic chemistry.
Introduction - History
Carbenes have thus played an important role in organic chemistry ever since the first
firm evidence of their existence. While much of the early chemistry was established in the 1950s
by Skell, it was Fischer’s group that introduced carbenes into inorganic and organometallic
chemistry in 1964, where they have been used in organic synthesis, catalysis, and
macromolecular chemistry. Briefly after the discovery of the first metal-carbene complexes (type
1), two reports by Ofele (Technische Universitat Munchen) and Wanzlick et al (Technische
Universitat Berlin) appeared that described complexes 2 and 3 with N-heterocyclic carbenes as
ligands.
1
Herrmann, W.A. et al. N-Heterocyclic Carbenes. Agnew. Chem. Int. Ed. Engl. 1997, 36, 1047.
CO
CO
CO W
CO
CO
R
Cr(CO)5
N
Ph
N
Ph
Hg
OMe
Ph
1
N
N
2
N
Ph
N
3
These unusual complexes were both obtained from imidazolium salts and metalcontaining precursors of sufficient basicity to deprotonate the organic substrate. In the first case,
a carbonylmetalate was used, while the Hg complex resulted from the metal acetate:
N
N
[HCr(CO)5]N
heat
-H2
Cr(CO)5
N
(1)
Ph
Hg(OAc)2, heat
N
-2AcOH
Ph
N
N
Hg
(ClO4)-
(2)
Ph
N
N
N
Ph
Ph
Ph
2ClO4-
In 1991, Arduengo et al succeeded in forming the first free, isolable, N-heterocyclic carbenes2:
2
Arduengo, A.J., et al. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113. P. 361-363.
R
R
N
N
CH X
NaH, cat. DMSO anion
N
R
-NaX, -H2
N
R
(3)
Deprotonation of the imidazolium ion is effected by NaH and catalytic amounts of KotBu or the
DMSO anion. One must note the limitations with regard to the substituent R, where Arduengo’s
work proved that a carbene of type 4 is thermodynamically stable. It is stable in the absence of
oxygen and moisture. Recrystallization from toluene affords clear, colorless rectangular prisms
with a melting point of 240-2410C. The 1H NMR spectral data in benzene-d6 are as follows:
δ 1.58 ppm (s, Ad, 12H), 2.01 (s, Ad, 6H), 2.29 (s, Ad, 12H), 6.91 (s, NCH, 2H).
The carbene above enjoys both steric and electronic stabilization. The electronic
stablization factors include a pi-donation into the carbene out-of-plane p orbital by the electronrich pi-system (:N-C=C-N:) and a good sigma-electronegativity effect. The pi-interactions lead
to several good resonance contributors for the carbene in which positive charge is delocalized in
the imidazole ring with the C2 represented as a pi-bonded carbanionic center, an interaction
useful in stabilizing nucleophilic carbenes.
Additional electronic stability for the carbene electron pair may be gained from the
sigma-electronegativity effects of the nitrogens on the carbene center. It was not the electronic
factors alone that allowed the isolation of the nucleophilic carbene, but also the steric hinderance
afforded by the adamantyl substituents (kinetic stability). This will be discussed in more detail
later on in the report.
Herrmann et al. then synthesized the crystalline divalent germanium(II) congener 5 form
diimine tBuN=CH-CH=NtBu, lithium, and germanium(II) chloride (will be discussed in the
latter part of the report).
As a result of this discovery, Denk et al. and Lappert et al. successfully synthesized the
stable silylenes 6 by reduction of the corresponding silicon(IV) cyclodiamides with K.
Compound 5 proved to be a successful precursor for chemical vapor deposition (CVD) of
amorphic germanium.
4
5
Ad
N
C
N
6
t-Bu
t-Bu
N
N
Ge
N
Si
N
t-Bu
t-Bu
Finally, Herrmann et al. suggested that N-heterocylic carbenes 4 would exhibit ligand
Ad
properties similar to those known form electron-rich phosphanes, leading to the conclusion that
an equally rich coordination and catalysis chemistry should arise from such C-nucleophilic
compounds.
Introduction – Hybridization & Structure
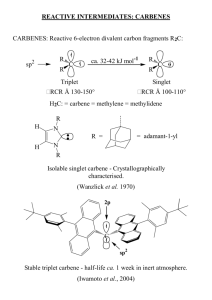
Even though isolable carbenes have been an interest for many years now, almost no
efforts were made to form them. Instead, stable “ylidene carbenes” have only recently been
isolated, as as the dihalocarbenes of sythetic chemistry. Carbenes are uncharged compounds with
a divalent carbon atom and two unshared electrons, which can be assigned to two nonbonding
orbitals in different ways. Thus, the chemistry of carbenes, nitrenes, and arynes are similar.
The electronic structure of a given carbene depends on its spin multiplicity and ground
state in the following ways: if the carbene has a nonlinear structure with an sp2-hybridized
carbon atom, the structures of the singlet ground state and the triplet state are comparitive to that
of carbenium ions (CR3)+ and free radicals . CR3, respectively. For the triplet state and excited
singlet states, linear structures with sp-hybridized carbon atoms must be considered. Most
carbenes have a nonlinear triplet ground state; however, carbenes with oxygen, sulfur or nitrogen
substituents as well as singlet-state dihalocarbenes are an exception.
Singlet Carbenes – Heteroatoms at the Carbene Center
Heteroatom donor groups on a carbene leave the originally degenerate orbitals on the
carbon unequal in energy, thus enhancing the nucleophilicity of the carbon atom as well as its
thermodynamic stability. The singlet-triplet splitting correlates with increasing electronegativity
of the pi-donor substituents X and Y in carbenes of the type :CXY.
Warwick et al. recognized that aromatic resonance structures in unsaturated Nheterocyclic five-membered rings contribute to carbene stability. Hoffmann et al. pointed out
that the ground state of the carbene will be a singlet state if the energy difference between the pi
and sigma orbitals is greater than 2eV, and that this splitting may be achieved by interaction of
the unoccupied carbene p orbital with an aromatic system of (4n+2)pi electrons.
Note that the heteroatom donor groups enhance the nucleophilicity of the carbon atom
and thus the thermodynamic stability. So far, only singlet carbenes with two nitrogen atoms have
been isolated as crystalline compounds so far (“carbon diamides”). These singlet carbenes have
a asserted low-energy HOMO and a high-energy LUMO. Because of the lower electronegativity
of carbon, they are stronger electron-pair donors than amines. Since the amino groups are pi-
donating and sigma-withdrawing, 2,3-dihydro-1H-imidazol-2-ylidenes benefit from a “pushpull” effect.3
One can see from the structure of the stable, sterically unencumbered ylidenes (ie. the
ones synthesized in Chm393), that not kinetic (steric hinderance) but rather thermodynamic
stabilization (electronic structure) is the essential feature. The difference in electronegativity of
nitrogen and carbon accounts for the inductive sigma effect, which stabilizes the pair of unshared
electrons in the in-plane carbene orbital. Also, the unoccupied p orbital yields a pi-resonance
interaction, where the N atoms donate their lone pairs to the carbon. Although steric hinderance
does contribute to the stability of carbenes (ie. the adamantyl group vs tBu/Me), the electronic
structure of the molecule is of utmost importance.
Electronic Structure of N-Heterocyclic Carbenes - Delocalization4
According to Arduengo et al., the stability of X is due to sigma donation of charge to the
nitrogen atoms, whereas pi backdonation plays a minor role.
X
Y
N
C
N
H
N
C
N
H
One might be ready to say that N-C-N pi delocalization is of minor importance. However, it is
necessary for an aromatic type of delocalization in the 6 pi-electron system in X. Thiel et al.
3
Arduengo, A.J., et al. A Stable Diaminocarbene. J. Am. Chem. Soc. 1995, 117. P.11027.
Herrmann, W.A. & Kocher, C. N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. Engl. 1997,
36, 1047.
4
attributed the difference between X and Y to the larger singlet-triplet gap in X involving a higher
barrier to dierization.
Aromaticity
Significant stabilization for the 6 pi-electron system can be achieved with two nitrogen
atoms. Thus, two oxygen atoms, with higher electronegativity than N, causes less stabilization.
Thus, as long as 6 pi-electron delocalization is important, so is the N-C-N pi delocalization.
Studies show that backdonation dominates carbene stability. The significant stabilizing effect of
the double bond indicates the presence of a certain degree of cyclic electron delocalization;
however, this stabilization is small when compared to the stabilization afforded from N-C-N pi
delocalization.
To conclude, the stability of carbenes is derived from a combination of steric and
electronic factors. The major contribution to carbene stabilization is due to p pi -p pi electron
donation form the two nitrogen atoms to the carbene carbon atom. Studies show that the
unsaturated cyclic carbene X is about 20kcal mol-1 more stable than the saturated carbene Y.
Note that for preparative chemistry, kinetic stability is crucial.
N-Heterocyclic Singlet Carbenes
In the early 1960s Wanzlick et al. investigated saturated and unsaturated heterocyclic
carbenes, focusing on C-C-saturated molecules of type 9. Imidazole was a precursor of 9, and
was formed from the reaction of 1,2-dianilinoethane with chloral, undergoing thermal alphaelimination of chloroform. The product dimerized to form the electron rich olefin 8a as a
colorless crystalline compound (4) that was incorrectly described as a ‘monomeric carbene’.
Most importantly, Lappert et al. developed a general synthesis for metal-carbene complexed by
treatment fo the electron-rich olefins with transition metal complexes such as chloro-
tris(triphenylphosphane)rhodium(I), discovering that this rhodium complex catalyzes the
metathetical cleavage of electron-rich olefins (will be explained in a latter portion of the report).
(4)
Ph
N
H
N
CCl3
Ph
-CHCl3
Ph
Ph
N
N
N
N
Ph
Ph
8
8a
Wanzlick et al. recognized that aromatic resonance structures in unsaturated Nheterocyclic five membered rings contribute to carbene stability. Thus, free carbenes such as
1,3,4,5-tetraphenyl-2,3-dihydro-1H-imidazol-2-ylidene (10) were generated as shown below
(however, they never successfully isolated the carbenes):
Ph
Ph
Ph
N
N
Ph
H
KOtBu
CCl3
-KX, -HOtBu
Ph
Ph
N
C
N
Ph
Ph
10
(5)
Synthesis & NMR Spectroscopy
Since the times of Nef (1895), who failed to isolate methylene, many believed that the
isolation of carbenes was an impossibility. However, about a century later, Arduengo et al.
successfully synthesized the first stable, crystalline carbenes. 1,3-diadamantyl-2,3-dihydro-1H-
imidazol-2-ylidene was obtained as a colorless crystalline solid by deprotonation of 1,3diadamantylimidazolium iodide with NaH in the presence of catalytic amounts of DMSO anion
(3). For the deprotonation to occur rapidly, catalytic amount so either KotBu or the DMSO anion
have to be used, since NaH/KH and the imidazolium salt are insoluble in THF. Stoichiometric
amounts of KotBu may be used, but this forms the less volatile tert-butly alcohol instead of
hydrogen. The best solvent was liquid ammonia. Depronation with NaH or potassium amide
occurred rapidly in a homogeneous phase to provide carbenes not previously accessible (ie.
oxygen-, nitrogen-, and diarylalkylphosphino- carbenes).
Ad
H
Ad
H
N
N
CH
H
Cl
N
N
H
Ad
Ad
11
Compound 11 is surprisingly thermally stable (m.p. 240-241oC without decomposition), but
decomposes at room temperature in solution.
A simple two-step approach to thermally stable, alkyl-substituted N-heterocyclic
carbenes was reported by Kuhn et al. An example was the reduction of 1,3,4,5tetramethylimidazol-2(3H)-thion with K in boiling THF yielded analytically pure 1,3,4,5tetramethyl-2,3-dihydro-1H-imidazol-2-ylidene (12) after filtration and solvent removal.
(6)
12
Me
Me
N
C=S
Me
Me
Me
K, THF, heat
C
N
Me
N
Me
N
Me
12
The thiones were formed by condensation of N,N’-dialkylthioureas with 3-hydroxy-2butanone. Note that the thermal lability of 2,3-dihydro-1H-imidazol-2-ylidenes in solution is
dependant on the nature of the substituents R on nitrogen.
(Ylidenes)
1,3-Disubstituted ylidenes with aryl residues (p-tolyl, p-chlorophenyl, and 2,4,6trimethylphenyl; m.p. 157(decomp), 154 (decomp), and 153oC (decomp), respectively) or higher
alkyl groups (t-butyl, cyclohexyl, adamantyl) and 1,3,4,5-tetramethyl-2,3-dihydro-1H-imidazol2-ylidene (m.p. 109oC) are colorless crystalline solids. 1,3-dimethyl-2,3-dihydro-1H-imidazol-2ylidene is an oily liquid, and can be kept in the solid state at –30oC under an inert-gas
atmosphere.
1,3-Dicyclohexyl-2,3-dihydro-1H-imidazol-2-ylidenes do not totally decompose upon
heating at 160oC for a day. In solution, they are stable to 50oC for several hours, as is 1,1’-(1,2ethylene)-3,3’-dimethylbis(2,3-dihydro-1H-imidazol)-2-ylidene.
Thus, as long as the solid carbnes or their solutions are colorless, they are very pure and
contain no decomposition products. Note that slightly yellow solutions are considered NMR
spectroscopically pure.
Imidazolium salts, which are good solvents for two-phase catalysis, are electron-rich
heteroaromatic compounds. Thus, the 13C NMR shift of the N-C-N sp2 carbon (the soon-to-be
carbene center) appears at d (delta) = 137. The H4(H5) ring protons and the CH-acidic H2 proton
resonate at d= 7.9-7.3 and 8-10 respectively.
Note that free 2,3-dihydro-1H-imidazol-2-ylidenes are diamagnetic, and thus the carbene
atoms resonate at rather low field (d= 210-220 in [D8]THF), with little influence from the
substituents on nitrogen on the chemical shift. The H4(H5) ring protons resonate at d= 6.9-7.0.
However, in 2,3-dihydro-1H-imidazol-2-ylidenes with aryl residues forming a coplanar
arrangement, anistropic effects of the aryl substituents account for chemical shifts at d= 7.6-7.8.
Thus, deprotonation of the imidazolium salts to the corresponding 2,3-dihydro-1H-imidazol-2ylidenes causes a significant high-field shift of the H4(H5) ring proton resonances in the case of
N-alkyl substitution, indicating a different magnetic anisotropy of the ring system, consistent
with a decrease in the 6pi-electron delocalization.
NR2Ga(R)NR2 and NR2Al(R)NR2 were experimentally verified with 1,4-diazabutadiene
(DAB) ligands. They are either stabilized by intramolecular coordination of a hemilabile
dimethylamino side group or by dimerization. Alkyl- and aryl- substituted diyls :E[CH(SiMe3)2]2
and :E(2,4,6-tBu3C6H2)2 (E= Ge & Sn) are monomeric in solution. Note that stability and
dimerization or insertion reactions reach a maximum for N-heterocyclic ring systems containing
the DAB fragment.
Solid-State Structures
X-ray and neutron diffraction techniques were used to study the solid-state structures of
2,3-dihydro-1H-imidazol-2-ylidenes and other C-C saturated and acyclic carbenes. For the 2,3dihydro-1H-imidazol-2-ylidenes, the N-C-N angles at the divalent carbon atom are practically
identical (ie. around 102o for ylidenes with methyl, p-tolyl, or adamantyl substituents). These
angles are significantly smaller than the typical range known for imidazolium salts (about 109o).
The C2-N1(N3) bond length for 1,3-diadamantyl-2,3-dihydro-1H-imidazol-2-ylidene was 1.37Å,
while that of the imidazolium cation was 1.33Å. This information brought the conclusion that pi
delocalization is less pronounced in 2,3-dihydro-1H-imidazol-2-ylidenes than in imidazolium
cations.
C-C Saturated Carbenes
When the C-C bond is saturated, the singlet-triplet gap shrinks, since a five-center sixelectron (5c-6e) pi delocalization is no longer a stabilizing factor. In 1995, Arduengo et al.
succeeeded in synthesizing imidazolidine-2-ylidene (15), a stable and crystalline monomer of the
electron rich olefins:
(9)
Mes
N
CH
N
Mes
Mes
KH, THF
-KCl, -H2
N
Cl
N
Mes
15
Ylidine 15 and 2,3-dihydro-1H-imidazol-2-ylidenes were synthesized by deprotonation of
amidinium salts.
Triazole-Derived Carbenes
The first triazole-derived carbene, 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene,
was prepared at 800C at 0.1mbar by endothermic elimination of methanol from the
corresponding 5-methoxytriazole in the solid state. The N-C bond lengths are much shorter than
expected due to a strong interaction of the filled nitrogen 2p orbitals with the unoccupied carbon
2p orbital.
DFT (density functional theorty) calculations on the spin density of the most stable
radical anion suggested that the single electron is delocalized between the N2-C3 double bond
and the adjacent phenyl substituent. Ylidene 16 decomposes above 1500C, and 1,3,4-triphenyl-
4,5-dihydro-1H-1,2,4-triazol-5-ylidene was the first carbene to be commercially available
(ACROS).
Ph
N N
Ph
N
Ph
CH
Ph
N N
NaOMe, MeOh
-NaClO4
C
Ph
N
16
Ph
(10)
Multidentate Carbenes
N-heterocyclic carbenes form a multitude of metal complexes, either over carbenes
formed by deprotonation of CH-acidic imidazolium salts, or in situ deprotonation by metalbound basic ligands (ie. amides, acetate, etc.). As two-electron ligands, carbenes are related to
ethers, amines, isonitriles, and phosphanes with regard to their coordination chemistry. Due to
the rigid geometry of the planar five-membered rings, the optimum number of atoms forming an
appropriate chelate ligand does not match that found with the diamines or diphosphanes.
Upon metal complexation, methylene-bridged 2,3-dihydro-1H-imidazol-2-ylidenes and
1,2,4,-1H-triazol-5-ylidenes form six membered rings. The natural bite angle of 780 in transition
metal complexes (W, Pd) of these ligands is exactly that of 1,1’-bis(diphenylphospino)methane,
a four membered ring. At rhodium centers, this dicarbene forms a bridging coordination mode.
A homoleptic hexacarbene-iron complex resulted after reaction with FeCl2. As well,
another tridentate carbene ligand, [1,3,5-{tris(3-tert-2,3-dihydro-1H-imidazol-2ylidene)methyl}-2,4,6]-trimethyl benzene] (20) was isolated as a solid.
Photoelectron Spectroscopy: A Study and Comparison of Carbenes, Silylenes, and Germylenes5
5
Arduengo, A.J., et al. Photoelectron Spectroscopy of a Carbene/Silylene/Germylene
Series. J. Am. Chem. Soc. 1994, 116, 6641.
Arduengo et al. investigated the photoelectron spectra for a whole series of isolated,
cyclic 6 pi-electron carbenes, silylenes, and germylenes. Density function calculations used to
explain the experimental data concluded that in contrast to the Si and Ge homologues, the
lowest-energy ionization process is the removal of an electron from the in-plane lone-pair orbital
which appears as the HOMO in the calculation.
The electronic structure of stable carbenes, silylenes, and germylenes in the imidazol-2ylidene have been studied via X-ray/neutron diffraction techniques, and are very interesting to
compare:
tBu
H
N
N
N
C
H
tBu
H
tBu
H
N
Si
H
N
N
tBu
tBu
tBu
1a
Ge
H
1b
1c
Photoelectron spectroscopy has been recently used by Denk et al. to study the energetics
of electrons in compounds containing the lighter main group IV elements in two-coordinate
oxidation state II compounds.
The series of compounds 1a-1c shows a smooth structural change as the two-coordinate
center is changed from C to Si to Ge. The N-E (E = C, Si, Ge) bond distances increase, and the
N-E-N angle steadily decreases. The carbene, silylene, and germylene were augmented in the
local minimum with the tert-butyls staggered, since this provides an average environment for
the two coordinate main group IV center.
The carbene 1,3-di-tert-butylimidazol-2-ylidene was found to have a HOMO (highest
occupied molecular orbital) that is basically the in-plane lone pair of electrons at the carbene
center (C σ-1p). The second ionization from the carbene occurs from a π-molecular orbital (π-3)
that is largely the C=C double bond in the imidazole ring with some contributions from the
nitrogens and the carbene center.
The HOMOs of the silylene and germylene are derived from the π-3 orbital, which
changes character to become more concentrated on the two-coordinate main group IV center (Si
or Ge) and less involved with the C=C double bond. Thus, the silylene and germylene structures
do not represent the type of bonding in the carbene 1a, but rather a degree of ‘zero-valent
chelated atom’ character.6
NHCs as Nucleophilic Catalysts (Synthetic Chemistry & Nature)
One of the most important uses of NHCs (N-heterocyclic carbenes) is their catalytic
applications in synthetic chemistry as well as in nature (eg. vitamin B1 enzyme cofactor thiamin
(22), a naturally ocurring thiazolium salt, plays a key role in biochemistry). Thiamin diphosphate
catalyzes the decarboxylation of pyruvic acid to active acetaldehyde as well as the benzoin
condensation of benzaldehyde. In basic aqueous buffers, the active catalyst of this reaction is a
2,3-dihydrothiazol-2-ylidene of the form 23.
OH
S
R
C
23
N
S
R
N
N
H3C
6
22
N
Arduengo, A.J., et al. Photoelectron Spectroscopy of a Carbene/Silylene/Germylene Series. J.
Am. Chem. Soc. 1994, 116, 6641.
2,3-Dihydro-1H-imidazol-2-ylidenes and 4,5-dihydro-1H-1,24-triazol-5-ylidenes are also
used catalytically in organic synthesis; for example, in the benzoin condensation of higher
aldehydes to alpha-hydroxyketones, the oxidative benzoin condensation of aldehydes, alcohols,
and aromatic nitro compounds to yield esters.
Nucleophilic ylidenes also serve as catalysts in the Michael-Stetter reaction:
R
R
N
CH
N
Et3N
C
R
R'CHO
S
S
N
R'
+
S
C OH
O
O
R'
O
The reaction yields 1,4-dicarbonyl compounds. Basically, a nucleophilic heterocyclic
ylidene attacks an aldehyde, and the generated acyl carbanion adds to the unsaturated ketones in
a 1,4 manner.
NHCs As Lewis Bases
2,3-Dihydro-1H-imidazol-2-ylidenes form stable adducts with soft and weak Lewis acids
such as iodine, in which the carbenes act as sigma donors. The nearly linear arrangement
suggests a hypervalent (10e-I-2c) central iodine atom (I-) with an expanded I . . . I bond and a
positively charged imidazole moiety of slightly pi delocalization. Thus, 2-iodo-1,3-
dimesitylimidazolium iodide reacted with 1,3-dimesityl-2,3-dihydro-1H-imidazol-2-ylidene to
give linear cationic bis(carbene) adducts of hypervalent iodine:
X= I, B(Ph)4
Mes
Mes
N
N
C-I
N
THF
I-C+
X-
N
Mes
Mes
Mes
C
+
N
N
X-
Mes
(17)
Upon complexation to a Lewis acid, electron density at C2 decreases, and ylidic
mesomeric structures become more important. 1,3-dimesityl-2,3-dihydro-1H-imidazol-2-ylidene
reacts with the corresponding imidazolium salt, a linear 3c-4e C-H-C hydrogen bridge is formed
(172.50). Note that this ‘proton complex’ has two different C-H bond lengths (202.60 and
115.90), perhaps due to the mesityl substituents’ favoring the formation of homoleptic
bis(carbene) adducts due to the cavity seen in the structure of the ylidene (19):
X= PF6, CF3SO3
Mes
Mes
Mes
N
N
N
CH
N
C
N
Mes
H-C
X-
N
Mes
THF
Mes
Mes
N
X
N
Mes
(19)
A new type of bimetallic complex was formed by cis-osmylation of the isolated double
bond of 1,3-dimethyl-2,3-dihydro-1H-imidazol-2-ylidene in [M(CO)5-(carbene)] (M= Cr, Mo,
W) with osmium tetraoxide (20). Upon oxidation, the ylidene becomes slightly more
nucleophilic.
M= Cr, Mo, W
Me
N
C-M(CO)5
N
OsO4, py
YP
Me
O
Os O
YP
O O
Me
N
C-M(CO)5
N
Me
(20)
Transition Metal Complexes
Transition metal complexes of NHCs are accessible by three different routes. Most
commonly, simple metal salts are treated with azolium precursors (route A), where the
coordination properties of the anion X- determine whether it enters the inner coordination sphere
of the resulting carbene complex. You may also do the reaction with free N-heterocyclic
carbenes (B), or perform the reaction with anionic metal-cyano complexes.
According to route B, nucleophilic N-heterocyclic carbenes may replace other ligands,
for example a two electron ligand (such as halide, CO, or acetonitrile) in the complex. From
carbonyl complexes such as [M(CO)6] (M= Cr, Mo, W), [Fe(CO)5], or [Ni(CO)4], one or two CO
molecules may be displaced.
R
R
N
X-
N
CH
N
C
N
R
R
A
[LnM(CN)]-, HBF4
C
multicomponent
condensation
B
H
R
RHN
N
M-Ln
N
N
M-Ln
N
R2
R
Synthesis of transition metal carbene complexes – A/B/C
Note that the range of NHCs which can be directly generated at metal centers is more
varied by route A due to the large variety of CH-acidic, cationic heteroaromatic compounds:
R
R
R
CH
CH
CH
N
N
N N
N
Imidazole
R
N
triazolium
R
N
benzimidazolium R
Heteroaromatic azolium salts which can be deprotonated in situ to give coordinated NHCs
C-deprotonation of azolium salts (as well as for triazolium, tetrazolium, and benzimidazolium
salts) is perfomed via anionic carbonylmetalates; examples are Wanzlick’s homoleptic Hg(II)
carbene complex and Ofele’s Cr(0) complex. Pd(0) complexes were also prepared via the metal
acetate:
Me
Me
N
I
N
Pd
N
heat
N
2
I-
CH
N
Me
n
(25)
Me
Cis/trans
Me
Me
N
I
+ Pd(OAc)2
NaI, 2KOtBu
Me
Me
N
I
Pd
I
N
Me
(26)
N
Pd
N
Me
A variety of rhodium and iridium complexes were also synthesized from azolium salts.
Carbene-complex formation is exclusively effected by proton transfer to the metal-bound ethoxy
group. Mono- and diylidene complexes of Rh and Ir are accessible a) directly from metal
alkoxides and azolium salts or b) from free N-heterocyclic ylidenes and dinuclear μ-chloro
complexes.
Nickel(II) Complexes of N-Heterocyclic Carbenes7
Since the discovery of free imidazol-2-ylidenes as stable, isolable reactants, their
importance as metal-attached ligands representing a new structure principle for homogeneous
catalysis has grown enormously. Although Ni(0) derivatives of NHC ligands have been
synthesized, Ni(II) complexes were not yet formed. Stable Ni(II) carbene complexes could act as
potential catalysts in CC-coupling reactions featuring a transmetallation step such as Grignard
cross-coupling or Suzuki coupling, which are extensively used in organic synthesis. The new
carbenes were expected to perform better than the traditional phophine-nickel(II) or phosphinepalladium(II) catalyst systems in terms of solubility, long term stability, and activity.
C6H11
N
2:
X2NiL2 +
a
C6H11
L= PPh3,THF
X= Cl, Br
R
rt, THF
-2L
N
N
X2Ni
-2HOAc
E
2
N
R
Me
2
N
H
E
N
I-
b
1a: X= Cl, R=C6H11, E=CH
1b: X= Br, R= C6H11, E=CH
1c: X= I, R= CH3, E= CH
2: X= I, R= CH3, E=N
Me
Preparation of Ni(II) carbene complexes from precursors by reaction with free carbenes or in situ by deprotonation
of azolium salts8
Herrmann, W.A. et al. Nickel(II) Complexes of NHCs. Organometallics. 1997. 16. P. 2209.
All bis(imidazol-2-ylidene)nickel(II) complexes 1a-c and 2 were isolated as violet-red solids of
which 1a-c are stable to air and soluble in polar organic solvents (ie. Chloroform, DMSO). 2 can
be dissolved in THF and heated to 240oC without decomposition.
Compounds 1a-c and 2 exhibit characteristic 13C NMR carbon shifts at δ 170 ppm (1a,b
in C6D6 and 1c CDCl3) and δ 187.1 ppm (2 C6D6). This resembles a clear upfield shift
compared to Ni(0) complexes of 20 ppm in the case of Arduengo’s NiL2 (L= 1,3dimesitylimidazol-2-ylidene). In the first catalytic experiments, compounds 1a,b proved their
suitability as catalyst precursors for the Suzuki coupling of phenylboronic acid with pchloroacetophenone.9
Main Group Element Metal Complexes
Monomeric and dimeric 1:1 adducts of Mg+2 and Zn+2 were obtained from the reaction of
diethyl zinc and diethyl magnesium with 1,3-diadamantyl and 1,3-dimesityl-2,3-dihydro-1Himidazol-2-ylidene:
R
N
(21)
C
N
R
M(et)2
M=Mg, Zn
R
et
+ C M
et
N
N
R
Lad, Lmes
The solid-state structure of the diethyl zinc adamantyl compound showed a trigonal-planar
coordination at zinc. The angle between the coordination sphere of zinc and the imidazole plane
8
9
Herrmann, W.A. et al. Nickel(II) Complexes of NHCs. Organometallics. 1997. 16. P. 2209
Herrmann, W.A. et al. Nickel(II) Complexes of NHCs. Organometallics. 1997. 16. P. 2209
is 81.60. However, mesityl groups do not provide as much steric bulk as adamantyl groups, as
shown by the diethyl magnesium mesityl compound. Thus, the 13C NMR of the imidazole C2
nuclei are shifted upfield by about 25 ppm.
The metal-donor bonds are mostly ionic and more labile in complexes of the heavier
analogues of Be and Mg. The solubility and stability of 2,3-dihydro-1H-imidazol-2-ylidene
complexes of Ca, Sr, and Ba decrease from Ca to Ba. The Ca and Sr adducts can be isolated in
the solid state at –360C, however, the Ba adduct dissociates.
B, Al, & Ga
Since NHCs do not depend on backdonation due to delocalization of the nitrogen lone
pair, 1:1 adducts with BH3 or BF3 are thermally stable and can even be sublimed without
decomposition. Both X-ray analysis and ab initio calculations on such borane adducts entail
alternating bond lengths and only weak pi interactions between the N-C-N moiety and the C-C pi
systems within the five-membered rings. Note that borane adducts are also known for N,O-
R
R
N
R
C MMe3
N
R
MMe3 M=Al,Ga
+
R=iPr/Me
R
R
R
C
N
+
N
C
N
+
BH2
R
R
R
N
Me2S*BH3 R
NMe3*AlH3
R
N
C
N
R
R
Et2O-BF3
AlH2
R=Mes,R'=H
R
heterocyclic carbenes. A silylene-borane adduct was recently reported.
N
C
N
+
BF3
R
Adducts of 2,3-dihydro-1H-imidazol-2-ylidenes and Lewis acids MR3 (M= Al, Ga; R=H, Me), BH3, and BF3
Catalysis
N-heterocyclic carbene complexes show great properties for a number of catalytic
reactions in organic chemistry, especially since imidazolium salts are cheap to synthesize for
industrial applications. N-heterocyclic carbenes have been overlooked even though they are
catalytically relevant ligands in coordination chemistry, and probably will play a greater role as
ligands in metal-containing catalysts in the near future.
(Catalysts in Homogeneous Catalysis)10
Phosphane and phosphite ligands not only protect low-valent metal centers from
aggregation (stabilization effect), but also create coordination sites in dissociation equilibria at
which the catalytic elementary steps proceed. Of industrial importance are the hydrocyanation
and hydroformylation reactions (Co1/Rh1/PR3). However, an excess of ligand (about 100 times
more than the metal) is needed, causing high running costs of technical plants. Phosphane and
phosphite complexes are also often water and air sensitive. Thus, new methods (ie. for the Heck
coupling reactions) are being used – relying on the special ligand properties of NHCs, mainly
their simplicity and efficiency. The new catalyst has three main advantageous properties and
potential for development: 1) high thermal and hydrolytic durability due to the extremely stable
M-C bonds. Thus, it has a long shelf-life and stability to oxidation. 2) It is easily accessible, and
3) there is no need for excess ligand.
The carbene-Pd complexes 1 and 2 (below) of the imidazole series were prepared from
Pd(OAc)2 and 1,3-dimethylimidazolium iodide in more than 70% yield. They are very stable to
heat, oxygen, and moisture. Catalyst 1 melts at over 2990C with partial decomposition and resists
Herrmann, W.A., et al. Metal Complexes of NHCs – A New Structural Principle for Catalysts
in Homogeneous Catalysis. Angew. Chem. Int. Ed. Engl. 1995, 34. No. 21.
10
treatment with O2 in boiling THF. Complex 2 melts at 280oC, but decomposes in solution (N,Ndimethylacetamide) at about 70oC.11
(a)
Me
I
N
N
H
2
(b)
Me
Me
I+ Pd(Oac)2
Pd
N
N
Me
Me
Me
Me
IH
+ Pd(Oac)2
Me
I
I
CH2
1
Me
N
Pd
N
N
N
N
I
N
N
N
C
H2
2
N
H
N
Me
Heck Olefination
The application of palladium complexes [PdL2I2] as catalysts for the Heck olefination of
aryl halides (39) is a major field of research. The catalysts are very stable to heat, moisture, and
oxygen, and they become highly active after a short induction period, making the carbene
complexes extremely suitable for the activation of chloroarenes.
Herrmann, W.A., et al. Metal Complexes of NHCs – A New Structural Principle for Catalysts
in Homogeneous Catalysis. Angew. Chem. Int. Ed. Engl. 1995, 34. No. 21.
11
R
+ H2C=CH-CO2nBu
cat
R
X
HC=CH-CO2nBu
One of the main area of interest to scientists such as Herrmann is the activation of
deactivated bromo- and chloroarenes for Heck-type reactions. For the past four years, scientists
have studied the efficient application of palladium carbene complexes with chelating ligands in
the Heck and Suzuki coupling of bromo- and chloroarenes. Herrmann et al. investigated the
Heck reaction with non-chelating carbene ligands, while recently, their attention has been
focused on the increasing importance of unsymmetrically substituted biaryl derivatives, which
are used as drug intermediates and nonlinear optical materials. Thus, it is of great interest to
investigate the transferability of the catalytic properties of Pd(II) carbene complexes to the
Suzuki coupling of aryl bromides with arylboronic acids:12
The palladium(II) complexes were formed using DMSO, not tetrahydrofuran, due to a higher
boiling point and higher solubility for all the reactants. This also allowed the chelating behaviour
of the ligand to be determined via X-ray diffraction for the first time:
12
Herrman, W.A. et al. Chelating NHC ligands in palladium-catlayzed heck-type reactions. J. of Organometallic
Chemistry. 557. 1998. 93-96.
Palladium (0) Complexes via Suzuki Cross Coupling13
The recent development of highly active palladium (0) catalysts for the activation of
chloroarenes in Heck-type reactions has been aimed on sterically demanding, basic, monodentate
phosphines. N-Heterocyclic carbenes (NHCs) are easily accessible resemble trialkyl phosphines
in that they are strongly -donating. Thus, homoleptic NHC-palladium(0) complexes are
catalytically active species, and their preparation is highly desireable.
There are various applications for these species, such as the versatility of NHC-ligands
for palladium(II) pre-catalysts in Heck-type reactions, the application of NHC-palladium(0)
olefin complexes in iodoarene Heck reactions, and the use of imidazolium salts in combination
with a palladium(0) source for a highly efficient Suzuki cross-coupling reaction. The Suzuki
cross-coupling reaction is a powerful tool for the preparation of bis-arenes, where the 14-electron
palladium(0) compounds are the catalytically active species.
(Synthesis)
Although homoleptic Ni(0) and Pt(0) complexes of NHCs could be prepared in solution,
this is not the case for the corresponding palladium(0) complexes. Even though the cocondensation of the free 1,3-di-tert-butylimidazolin-2-ylidene (1b) and palladium vapor has
successfully led to the desired complex 3b in 32% yield, a more general and efficient method is
to prepare these complexes by ligand exchange.
Basically, the free NHC is added to a slurry of bis(tri-ortho-tolylphosphine) palladium(0)
in toluene at room temperature to form a clear yellow solution. Complex 3 was isolated
analytically pure in over 60% yield after washing away the liberated tri-ortho-tolylphosphine
with cold n-hexane. The ligand exchange was monitored by 31P-NMR to reveal that full
conversion occurred in all cases after addition of an excess of NHC. Any loss of product occurs
during the work-up. NMR data shows that the reaction occurs via a step-wise exchange of the
phosphine ligands:
13
Bohm, V.P.W. et al. N-Heterocyclic Carbenes. Part 26. Journal of Organometallic Chemistry. 595. 2000. P.
186-190.
R
N
C
N
R
R
(o-tol)3P-Pd-P(o-tol)3
1
toluene
RT
R
2
N
C-Pd-P(o-tol)3
N
R
1
toluene
RT
R
N
N
Pd
N
N
R
R
R= mes (a), t-Bu (b), i-Pr (c), cyclohexyl (d).
13
C-NMR spectra show that the carbene carbon signals are shifted approximated 20ppm
to higher field upon complexation to palladium(0). In comparison to related palladium(II)
complexes, the signals are shifted about 30ppm to lower field. Complex 3a is stable towards
reaction conditions of the catalysis experiments, whereas complexes 3b – 3e readily decompose
to palladium black. All complexes (except for 3c) are moderately solube in most organic
solvents.
Olefin Metathesis
Ruthenium(II) carbene complexes were successfully used as catalysts in olefin
metathesis. The bond between ruthenium and the NHC carbon atom does not react with strained
cyclic olefins; thus, the carbene acts as a stabilizing “spectator ligand”. Therefore, Ruthenium(II)
complexes with NHCs are similar to analogous complexes with basic phosphanes (eg. [Ru(pcymene)Cl2(PCy3)3]) in their catalytic performance.
Thus, we have established the fact that NHCs complement and extend the capabilities of
the ubiquitous phosphanes. In olefin metathesis, ruthenium alkylidene compounds 2 bearing two
NHC ligands exhibit a catalytic activity comparable to that of the phosphane system 1:14
Thus, it is a combination of NHCs with coordinatively more labile ligands on the ruthenium
center that allows NHCs to develop their full potential as catalysts. In the catalytic cycle of olefin
metathesis the mechanistic scheme for 1 postulates the dissociation of a phosphane ligand as the
key step in the dominant reaction pathway. The strong metal-NHC bond raises the question as to
whether this mechanism would work for metathesis catalyst 2.
By calculating the dissociation energies of NHC and phosphanes for ruthenium-alklidene
model compounds according to DFT (density functional) methods, scientists demonstrated that
the ligand dissociation energies ascend n the series PH3<PMe3<NHC. Due to the higher
coordination energy, the dicarbene complexes 2 should disfavor a dissociative pathway similar to
that of 1. To solve this problem, complexes 3 were formed by the addition of 1.2 equivalents of
the appropriate NHC to a solution of 1 in THF (low temperature is necessary).
14
Gleich, Dieter et al. Highly Active Ruthenium Catalysts for Olefin Metathesis: The Synergy of NHCs and
Coordinatively Labile Ligands. Angew. Chem. Int. Ed. 1999. 38. No. 16.
At room temperature, the selectivity is lower and mixtures with significant amounts of
the dicarbene complexes 2 were generated. This illustrates the stablity of the complexes as well
as the higher Lewis basicity of NHCs with respect to trialkylphosphanes.15
To conclude, the synonymous nature of NHCs and coordinatively labile ligands allows
for the synthesis of catalysts for olefin metathesis that combine high catalytic activity with
excellent stablity against air and moisture. This concept also applies to other catalytic processes,
such as the palladium-catalyzed coupling reactions.
Chemical Vapour Deposition (CVD) of Germanium16
Photoelectron spectroscopy (PES) and thermolytic studies opened an important
application of the CC-saturated cyclogermylene B in material science. The compound yielded
[Ge/GeH] layers, A, and N,N’-bis-butylethylenediamine upon heating to 470K.
15
16
Ibid.
Herrmann, W.A., et al. Stable Cyclic Germanediyls. Angew. Chem. Int. Ed. Engl. 1992, 31, No. 11. P. 1485.
A CVD study showed that these cyclogermylenes are excellent precursors for generating
thin films of amorphic α-germanium upon sublimation at about 40oC/0.25mbar. Germanium
coatings start growing at about 245oC. At lower temperatures selective deposition of germanium
on silicon patterned with silicon dioxide is possible! To show the extreme selectivity, silicon
wafers covered with 1.8μ thick patterned SiO2 were used, and the contact holes were filled at a
substrate temperature of 2300C with Ge without any deposition onto the SiO2 surface.
The decomposition of cyclogermylene B occurs via a bimolecular mechanism where one
germanium atom is deposited, while an equivalent amount of the CC-unsaturated derivative A is
formed together with a diamine.
The arsenic-cobalt heterobimetallic compound C forms thin films of cobalt arsenide under CVD conditions
at low temperatures, yielding uniform films with crystalline and electrical properties. Impurities due to carbon,
oxygen, and nitrogen are usually below 1%.
Germylenes as a source for depositing alpha-Ge films (next page)
tBu
tBu
twist
N
tBu
N
Ge
N
tBu
A
planar
N
N
tBu
C
heat
heat
amorphous alpha-Ge
2
Ge
N
tBu
CoAs
tBu
But
tBu
N
As Co(CO)4
Ge
N
tBu
B
N
NH
470K
+
"Ge" +
NH
Ge
N
But
tBu
Germanium Dichloride Dioxane Complex17 (A Base-Stabilized Germylene)
Dicoordinate germanium compounds (“germylenes”) are highly reactive compounds
discovered recently by M.F. Lappert in 1980. They can be considered bigger and more stable
than most carbenes, exhibiting the trend that the heavier members of the group are more stable in
lower oxidation states.
Germylenes have both electrophilic and nucleophilic reactivity as one might expect from
the electronic structures shown below, featuring a nucleophilic lone pair and an electrophilic
17
Chm393 lab manual - Germanium experiment & Chm331 website.
empty orbital. Although silylenes and carbenes only occur as singlets, other carbenes could have
a triplet ground state.
Germylenes are highly air sensitive and can only be handled under inert gas, with the
exception of GeI2 and GeCl2 (1,4-dioxane), which are fairly air stable, and thus are useful
starting reagents in many reactions. The unusual stability of these compounds is due to the
coordinative saturation of the germanium center. GeI2 is stabilized in an extended polymeric
structure by the soft and polarizable iodide ions. Chloride ions are apparently not soft enough to
adopt the same lattice, and thus exists as the polymeric compound. GeI2 has the CdI2 structure in
which the metal reaches coordination number 6 (octahedral). This structure is not very ionic and
has much covalent interactions between the cations and anions. With Cl- as the counterion,
Ge(II) does not form a three-dimensional lattice, but polymerizes to form covalent chains
composed of GeCl2 units, which is unreactive and insoluble in all solvents, rendering it useless
as a starting material.
GeCl2 can be stabilized by the coordination of the 1,4-dioxane as Lewis base, resulting in
the adduct GeCl2 (1,4-dioxane), a colorless crystalline compounds that is fairly stable in air. It
could be stored indefinitely and is a very good starting material for germanium chemistry.
Although not commercially available, it is easily formed by the reduction of GeCl4:
GeCl4 + Et3SiH + 1,4-dioxane ----reflux---> [GeCl2<--1,4-dioxane] +Et3SiCl + HCl
HCl is a byproduct of the reaction, and must be ventilated adequately to prevent pressure build
up. Note that GeCl4 is relatively water sensitive and thus the entire reduction is carried out under
inert gas. It is important to note the following prior to attempting the experiment:
GeCl4 (US $217/100g) is very expensive and extremely air and water sensitive, attacking
most greases used in the lab. Thus, it is stored in a special bottle with a teflon valve, or in
a small sealed glass bottle.
1,4-dioxane is cancerogenic, thus USE THE FUMEHOOD ALWAYS!
LiAlH4 is very moisture sensitive, and thus must be handled in dry conditions only. Also,
always add the LiAlH4 to solvents, not the other way around.
The high price of germanium made it necessary to have high yield procedures for the synthesis
of germanium compounds. Thus, the GeCl2 (1,4-dioxane) could be achieved by a number of
ways:
(1)
Use of a furnace and the flow of HCL gas needs to be constantly monitored. The reaction
is slow, and yields only about 50% product.
(2)
Use of highly toxid tri-n-butyl tin hydride as reducing agent, giving about 80% yield and
completely separating the product from the side product tributyl tin hydride.
(3)
The method using the inexpensive triethylsilane as catalyst, usually yielding about 90%
product.
1,3-Di-tert-butylimidazolium chloride and 1,3-Di-tert-butylimidazole-2-ylidene were
subsequently synthesized, and a thorough study of N-Heterocyclic carbenes ensues in the
‘Discussion’. The chemical reactions are shown in the ‘Discussion’ as well.
Synthesis of GeCl2 (1,4-dioxane) – Chm393 laboratory
All operations were conducted under nitrogen and with dried solvents. 10mmol GeCl4
and 20mmol triethylsilane were dissolved in 15mL of 1,4-dioxane, with the addition of LiAlH4
catalyst. This mix was boiled to relux for over four hours. Crystallization of the product was
completed by cooling in an ice bath for a short period of time (so the solvent does not freeze as
well). The solvent was removed with a syringe and disposed into the appropriate germanium
waste bottle. The white crystalline product was washed with 10mL of dry 1,4-dioxane and dried
under dynamic vacuum for 10 minutes to yield 1.30g (90.59% yield).
Sublimation of 1,3-Di-tert-butyl-3,5-diaza-1-germole
tBu
N
2 Li
THF
N
tBu
tBu
tBu
N Li
N Li
GeCl2(dioxane)
-2LiCl
THF
tBu
N
Ge
N
tBu
A solution of 1,4-di-tert-butyl-1,4-diazadiene (5.94 mmol) in 50mL THF was reduced
with 12mmol lithium wire until the solution turned red. The red solution was added to
GeCl2(1,4-dioxane) to form a brown solution, which was stirred at room temperature for an hour.
The solvent was removed under vacuum, and the residue dissolved in hexane and filtered
through a schlenk frit.
Last year, the 393 class failed to complete the experiment. Thus, we attempted to sublime
their product (stored in an air-tight seal under inert gas), but failed to produce any of the
colorless pure germylene (sublimed, but decomposed)! It was a good learning experience though,
as we learned how to sublime chemicals properly.
FT-IR (Nujol, cm-1) v = 1466s, 1364s, 1253m, 1217s, 1158m, 1056m, 813w, 721m,
697s, 578m, 505s, 481w, 446w, 422m.
1H-NMR (C D ) δ = 1.43 [s, CH ], 7.05 [s, =CH ]
6 6
3
2
13C-NMR (C D ) δ = 33.2 [q, 1J(C,H) = 125.1], 55.7 [s, CMe ].
6 6
3
Synthesis of 1,3-Di-tert-butylimidazolium chloride
tBu
12M HCl
N
tBuNH2 + (CH2=0)n
Glyoxal
+
C H
N
Cl-
tBu
In a 100mL 3-neck flask with a condenser, thermometer, and an addition funnel, 21mL of
tert-butylamine (0.2 mol) were slowly added to 3g of paraformaldehyde (0.1g). The reaction
temperature was not allowed to exceed 45 degrees Celcius. Once at room temperature, 8.33mL
of HCl (0.1mols) were added over a very short time period (about 15 minutes), yielding a white
precipitate. 14.5mL of 40% glyoxal (0.1 mol) was then added, yielding a deep red solution,
which was refluxed for one day at 120 degrees Celcius.
Water was removed under reduced pressure using a rotary evaporator, yielding an oily
and sticky brown product (92% yield). The carbenium salt was purified by washing with much
acetone yielding the white solid, but this was a slow and tedious process. After washing with
acetone, we only ended up with 58% yield. An 1H NMR was taken of the product in deuterated
chloroform, and was found to contain water. However, the expected peaks at 1.778 (singlet, due
to t-Bu groups) and 7.661 (due to protons on the double bond) appeared. The peak at 10.36 did
not appear, however a peak at 9.888 appeared (I think the peak was shifted here due to
impurities). Small peaks at 5.928, 5.002, 4.183, and 8.309 appeared, but these are definitely due
to impurities (ie. water). A small percent of undeuterated chloroform may have formed the small
peak at 7.275.
1H
NMR
(CDCl3): δ = 1.80 ppm [s, 18H, t-Bu], 7.634 [s, 2H, CH], 10.36 [s, 1H, CH]
FT-IR
(NaCl; Nujol) 611sh, 639s, 723sh, 745s, 808br, 829sh, 913s, 977w, 1096m,
1124m, 1173s, 1209s, 1293s, 1377s, 1546s, 1961w, 2081w, 2418w, 2460w,
2509w, 2608m, 2678m, 2917s, 3156m, 3360m.
Synthesis of 1,3-Di-tert-butylimidazole-2-ylidene
tBu
tBu
N
nBuLi
+
C H
N
Cl-C4H10(g)
-LiCl(s)
tBu
N
C
N
tBu
1.86mL of n-butyllithium (2.5M, 4.65 mmol) was added dropwise to a 10mL THF
suspension of 1,3-Di-tert-butylimidazolium chloride (0.5g, 4.65mmol) in a 100mL two-neck
round bottom flask. The reaction was stirred for about two hours, and the suspension was (1)
filtered under the exclusion of air and (2) sublimed. The solvent was removed under reduced
pressure.
FT-IR
(NaCl); v = 2967 v, 1661v, 1474s, 1403br, 1263s, 1170m, 1077m, 926w, 812m.
13C {1H}
NMR (200MHz, C6D6): δ 31.46 ppm [q, t-Bu], 55.76 [s, t-Bu], 115.00 [d, CH=CH],
212.87 [s, N2C:]
(Germanium Complexes)
GeCl2 can be stabilized by the coordination of the 1,4-dioxane as Lewis base, resulting in
the adduct GeCl2 (1,4-dioxane), a colorless crystalline compounds that is fairly stable in air. It
could be stored indefinitely and is a very good starting material for germanium chemistry.
Although not commercially available, it is easily formed by the reduction of GeCl4:
GeCl4 + Et3SiH + 1,4-dioxane ----reflux---> [GeCl2<--1,4-dioxane] +Et3SiCl + HCl
GeCl4 is relatively water sensitive and thus the entire reduction is carried out under an
atmosphere of either Argon or Nitrogen. The whole reaction could be seen as a redox reaction, in
which Ge is reduced from +4 to +2.
(Stable Cyclic Germanediyls [’Cyclogermylenes’])18
When 1,4-di-tert-butyl-3,5-diazadiene in THF is reduced with Li wire and added to
GeCl2(1,4-dioxane), 1,3-Di-tert-butyl-3,5-diaza-1-germole is formed, as shown below:
L= 1,4-dioxane
tBu
tBu
GeCl4 +
1a
GeCl2*L +
-Et3NHCl
NH
tBu
2a
tBu
tBu
tBu
NLi -2 LiCl
N
2b
NLi
2c
18
tBu
Ge
3b
tBu
tBu
tBu
tBu
NLi
3b
+2 Li
-2 LiCl
N
NLi
1b
GeCl2*L +
Cl
Ge
N Cl
N
Et3N
NH
N
-2 LiCl
Ge
N
tBu 3c
Herrmann, W.A., et al. Stable Cyclic Germanediyls. Angew. Chem. Int. Ed. Engl.
1992, 31, No. 11. P. 1485.
Substituted ethylenediamine and its derivatives are generally suitable for the stabilization
of main group and subgroup elements with unusual valencies, especially germanium. The cyclic
germanium(IV) diamide 3a could be formed from GeCl4, and the diamine 2a in the presence of
triethylamine. Reductive dehalogenation of 3a provides the germanediyl (“germylene”) 3b.
Another way is to react the dilithium salt 2b with GeCl2(1,4-dioxane). The CC-unsaturated
germanediyl 3c is usually obtained as a side product of this reaction. 3c could also be obtained
purely by the treatment of the dilithium salt 2c with GeCl2(1,4-dioxane).
Reactivity
Both germanediyls are infinitely stable at room temperature under inert gas (as done by
last year’s class). The volatile compounds are easily hydrolyzed, and sublime at 45 degrees
Celcius. 3c is less reactive than 3b, and is more thermally stable, and can even be briefly handled
in air when crystalline. Germanediyls 3b and 3c are monomeric in both solid and gas phase, but
when the t-butyl groups are replaced by methyl groups, dimerization and trimerization occurs.
Structure
X-ray structure analysis of 3b shows the atoms of the C2N2Ge five-membered ring to lie
in a mirror plane. Calculations predict that the compound should have a twist conformation in the
ground state. Compound 3c is planar; however, there seems to be no electronic differences
between the two molecules.
Electronic Structure of N-Heterocyclic Germylenes19
Due to their similarity, silylenes and germylenes were studied in parallel with the
carbenes. Even though Arduengo et al. concluded that the electron-density distribution of
silylenes and germylenes differ significantly from that of carbenes, numerous thermodynamic
19
1047.
Herrmann, W.A. & Kocher, C. N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. Engl. 1997, 36,
calculations conclude that silylenes and germylenes are also stabilized by pπ-pπ delocalization.
6π-electron delocalization is shown to be present, but less pronounced than in the corresponding
carbene. The π-delocalization in N-heterocyclic carbenes will be examined further in this
discussion.
NHCs & the Future
Since free carbenes are now available through the work of Arduengo (1991), a rebirth in
this overlooked area occurred. The particular strength of these compounds is their universal
ability to coordinate to metal centers, varying from electron-rich transition metals (ie. Pd0) to
electron-poor main group metal cations (ie. Be+2), and high oxidation state metals such as TiIV,
NbV, and ReVII. Thus, NHCs are even more versatile than phosphanes in that their sigma
donation suffices to form stable adducts with certain metals, usually to form highly stable bonds
with catalytically relevent metals (ie. Pd0 carbene complexes for the Heck coupling reactions).
NHCs also withstand a variety of electronic situations – making them ideal for
homogeneous catalysis. Thus, side chains containing hemilabile P, N, and O should enhance the
catalyst’s stability.
NHCs are easily available as free compounds from the deprotonation of azolium salts in
liquid ammonia, thus making a overabundance of substituted derivatives (that are important in
chemical synthesis) available. NHCs are also well-matched with any type of main group and
transition metal element of the PTE, in nearly any oxidation state. NHCs also form highly
efficient catalysts with some metals (ie. Pd(II) complexes used in the Heck-type C-C coupling).
References
Alder, R.W., et al. Bis(diisopropylamino)carbene. Angew. Chem. Int. Ed. Engl. 1996. 35.
No.10.
Arduengo, A.J. et al. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991. 113. P. 361.
Arduengo, A.J. et al. A Stable Diaminocarbene. J. Am. Chem. Soc. 1995. 117. 11027.
Arduengo, A.J. et al. Electronic Distribution in a Stable Carbene. J. Am. Chem. Soc. 1994. 116.
p. 6812.
Arduengo, A.J. et al. Electronic Stabilization of Nucleophilic Carbenes. J. Am. Chem. Soc.
1992. 114. P. 5530.
Arduengo, A.J. et al. Photoelectron Spectroscopy of a Carbene/Silylene/Germylene Series.
J. Am. Chem. Soc. 1994. 116. P. 6641.
Artus, G.R.J. et al. Metal Complexes of N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. Engl.
1995. 34. No. 21.
Bohm, V.P.W. et al. N-Heterocyclic Carbenes. Part 26. Journal of Organometallic Chemistry.
595. 2000. P. 186-190.
Denk, M.K. et al. Nucleophilic Carbenes and the Wanzlick Equilibrium: A Reinvestigation.
Tetrahedron Letters. 1999, 40. P. 2057.
Gerstberger, G. et al. Nickel(II) Complexes of N-Heterocyclic Carbenes. Organometallics.
1997. 16. P. 2209.
Gilchrist, T.L. et al. Carbenes, Nitrenes, and Arynes. Lord Tedder. USA. 1969. Chp. 9.
Gleich, D. et al. Highly active Ruthenium Catalysts for Olefin Metathesis. Agnew. Chem. Int.
Ed. Engl. 1999. 38. No. 16.
Herrmann, W.A. et al. N-Heterocyclic Carbenes. Agnew. Chem. Int. Ed. Engl. 1997. 36. 2162.
Herrman, W.A. et al. Chelating NHC ligands in palladium-catlayzed heck-type reactions. J. of
Organometallic Chemistry. 557. 1998. 93-96.
Herrmann, W.A. et al. Stable Cyclic Germanediyls (“Cyclogermylenes”). Agnew. Chem. Int.
Ed. Engl. 1992. 31. No. 11.
Huang, J. et al. Olefin Metathesis- Active Ruthenium Complexes Bearing a Nucleophilic
Carbene Ligand. J. Am. Chem. Soc. 121. 2674.
Kirmse, W. Carbene Chemistry. Academic Press. New York. 1964. Page 5.
Regitz, Manfred. Nucleophilic Carbenes: An Incredible Renaissance. Angew. Chem. Int. Ed.
Engl. 1996, 35. No. 7.