PROT_22394_sm_suppinfo
advertisement

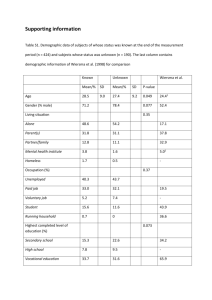
SUPPLEMENTARY MATERIAL FOR: Construct optimization for protein NMR structure analysis using amide hydrogen / deuterium exchange mass spectrometry Seema Sharma,1,2 Haiyan Zheng,1 Yuanpeng J. Huang,1,2 Asli Ertekin,1,2 Yoshitomo Hamuro,3 Paolo Rossi,1,2 Roberto Tejero1,2, Thomas B. Acton,1,2 Rong Xiao,1,2 Mei Jiang,1,2 Li Zhao,1,2 Li-Chung Ma,1,2 G. V. T. Swapna,1,2 James M. Aramini,1,2* and Gaetano T. Montelione1,2,4* 1 Center for Advanced Biotechnology and Medicine, Department of Molecular Biology and Biochemistry, Rutgers, The State University of New Jersey, 679 Hoes Lane, Piscataway, New Jersey 08854 2 Northeast Structural Genomics Consortium, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854 3 4 ExSAR Corporation, Monmouth Junction, New Jersey 08852 Department of Biochemistry, Robert Wood Johnson Medical School, University of Medicine and Dentistry of New Jersey, Piscataway, New Jersey 08854 Figure S1. Selected examples of Disorder prediction Metaserver1 results for proteins from the Northeast Structural Genomics Consortium, based on various protein disorder prediction software - DISEMBL,2 DISOPRED2,3 DISPro,4 DRIP-PRED,5 FoldIndex,6 FoldUnfold,7 GlobPlot2,8 IUPred,9 Prelink,10 RONN,11 VL2,12 VL3,13 VL3H,13 VSL214. Details about the Disorder prediction Metaserver and its parameterization will be presented elsewhere. (A) Disorder prediction Metaserver1 results for Spr lipoprotein from Escherichia coli, NESG target ER541. In this case the prediction programs provide a clear consensus result, namely strong evidence for disorder in the N-terminal region of the protein (red arrow). On the basis of these results, various truncated constructs lacking residues from this region were generated, ultimately leading to the production of Spr[37162] (green arrow) whose solution NMR structure was solved by the NESG consortium (PDB ID, 2K1G).15 (B) Disorder prediction Metaserver1 results for the SOS response protein YnzC from Bacillus subtilis, NESG target SR384. In this case no definitive boundary for the disordered region is obtained from the prediction programs. Consequently, we employed the DXMS technique to precisely design constructs lacking a disordered C-terminal tail (see main text). Figure S1A. DisMeta server results for E. coli Spr (NESG target, ER541). Figure S1B. DisMeta server results for B. subtilis YnzC (NESG target, SR384). Table S1: Deuterium uptake levels (10, 100, and 1000 sec incubation at pH 7.5 and temperature ~ 0o C) for the peptides selected for DXMS analysis of B. subtilis YnzC (NESG target SR384). Peptide selected 1-12 1-13 13-24 14-24 13-40 26-35 41-51 52-61 59-71 62-85 68-85 10 sec 2H level (%) 46 34 27 35 34 19 80 80 78 81 76 100 sec 2H level (%) 88 75 70 81 66 48 93 94 88 90 84 1000 sec 2H level (%) 90 92 86 93 90 97 93 86 97 91 92 Supplementary References 1. Huang YJ, Montelione GT. DisMeta disorder prediction Metaserver. Rutgers University. http://www-nmr.cabm.rutgers.edu/bioinformatics/disorder/ 2. Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure 2003; 11: 1453-1459. 3. Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 2004; 337: 635-645. 4. Cheng J, Sweredoski MJ, Baldi P. Accurate prediction of protein disordered regions by mining protein structure data. Data Mining and Knowledge Discovery 2005; 11: 213-222. 5. MacCallum RM. Order/Disorder prediction with self organizing maps. CASP 6 meeting. Online paper: http://www.forcasp.org/paper2127.html. 6. Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex©: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005; 21: 3435-3438. 7. Galzitskaya OV, Garbuzynskiy SO, Lobanov MY. FoldUnfold: web server for the prediction of disordered regions in protein chain. Bioinformatics 2006; 22: 29482949. 8. Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res 2003; 31: 3701-3708. 9. Dosztányi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005; 21: 3433-3434. 10. Coeytaux K, Poupon A. Prediction of unfolded segments in a protein sequence based on amino acid composition. Bioinformatics 2005; 21: 1891-1900. 11. Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005; 21: 3369-76. 12. Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein disorder. Proteins 2003; 52: 573-584. 13. Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins 2003; 53 (S6): 566-572. 14. Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics 2006; 7: 208. 15. Aramini JM, Rossi P, Huang YJ, Zhao L, Jiang M, Maglaqui M, Xiao R, Locke J, Nair R, Rost B, Acton TB, Inouye M, Montelione GT. Solution NMR structure of the NlpC/P60 domain of lipoprotein Spr from Escherichia coli: structural evidence for a novel cysteine peptidase catalytic triad. Biochemistry 2008; 47: 9715-9717.