PCR
advertisement

Partial Cox Regression Analysis
1. Literature Survey
1.1 Survival Analysis
Survival analysis is the branch of applied statistics dealing with the analysis of data on
times of events in individual life-histories (human or otherwise). A more modern and
broader title is generalized event history analysis. To begin with, the event in question
was often the failure of a medical treatment designed to keep cancer patients in remission
and the emergence and growth of survival analysis was directly connected to the large
amount of resources put into cancer research. This area of medical statistics brought a
whole new class of problems to the fore, especially the problem of how to deal with
censored data. At first many ad hoc techniques were used to get around these problems
but slowly a unifying idea emerged. This is to see such data as the result of a dynamic
process in time, each further day of observation producing some new pieces of
data. Tractable statistical models are based on modeling events continuously in time,
conditioning on past events; and new statistical ideas such as partial likelihood are also
based on this dynamic time structure.
Survival analysis examines and models the time it takes for events to occur. The
prototypical such event is death, from which the name ‘survival analysis’ and much of its
terminology derives, but the ambit of application of survival analysis is much broader.
Essentially the same methods are employed in a variety of disciplines under various
rubrics – for example, ‘event-history analysis’ in sociology. Survival analysis focuses on
the distribution of survival times. Although there are well known methods for estimating
unconditional survival distributions, most interesting survival modeling examines the
relationship between survival and one or more predictors, usually termed covariates in
the survival-analysis literature. The Cox proportional-hazards regression model
(introduced in a seminal paper by Cox, 1972), a broadly applicable and the most widely
used method of survival analysis. The survival library in R and S-PLUS also contains all
of the other commonly employed tools of survival analysis.
There are many texts on survival analysis: Cox and Oakes (1984) is a classic source.
Allison (1995) presents a highly readable introduction to the subject based on the SAS
statistical package, but nevertheless of general interest. Extensive documentation for the
survival library may be found in Therneau (1999).
1.2 Basic Concepts and Notation
Let T represent survival time. We regard T as a random variable with cumulative
distribution function P (t) = Pr(T ≤ t) and probability density function p(t)=dP (t)/dt. The
more optimistic survival function S(t) is the complement of the distribution function,
S(t) = Pr(T> t) = 1− P (t). A fourth representation of the distribution of survival times is
the hazard function, which assesses the instantaneous risk of demise at time t, conditional
on survival to that time:
Modeling of survival data usually employs the hazard function or the log hazard. For
example, assuming a constant hazard, h(t) = ν, implies an exponential distribution of
survival times, with density function p(t) = νe−νt . Other common hazard models include
which leads to the Gompertz distribution of survival times, and
which leads to the Weibull distribution of survival times. In both the Gompertz and
Weibull distributions, the hazard can either increase or decrease with time; moreover, in
both instances, setting ρ =0 yields the exponential model. A nearly universal feature of
survival data is censoring, the most common form of which is right-censoring: Here, the
period of observation expires, or an individual is removed from the study, before the
event occurs – for example, some individuals may still be alive at the end of a clinical
trial, or may drop out of the study for various reasons other than death prior to its
termination. An observation is left-censored if its initial time at risk is unknown. Indeed,
the same observation may be both right and left-censored, a circumstance termed
interval-censoring. Censoring complicates the likelihood function, and hence the
estimation, of survival models.
Moreover, conditional on the value of any covariates in a survival model and on an
individual’s survival to a particular time, censoring must be independent of the future
value of the hazard for the individual. If this condition is not met, then estimates of the
survival distribution can be seriously biased. For example, if individuals tend to drop out
of a clinical trial shortly before they die, and therefore their deaths go unobserved,
survival time will be over-estimated. Censoring that meets this requirement is
noninformative. A common instance of noninformative censoring occurs when a study
terminates at a predetermined date.
1.3 The Cox Proportional-Hazards Model
As mentioned, survival analysis typically examines the relationship of the survival
distribution to covariates. Most commonly, this examination entails the specification of a
linear-like model for the log hazard. For example, a parametric model based on the
exponential distribution may be written as
or, equivalently,
that is, as a linear model for the log-hazard or as a multiplicative model for the hazard.
Here, i is a subscript for observation, and the x’s are the covariates. The constant α in this
model represents a kind of log-baseline hazard, since log hi(t)= α [or hi(t)= eα] when all
of the x’s are zero.
The Cox model, in contrast, leaves the baseline hazard function α(t) = log h0(t)
unspecified:
or, again equivalently,
This model is semi-parametric because while the baseline hazard can take any form, the
covariates enter the model linearly. Consider, now, two observations i and i′ that differ in
their x-values, with the corresponding linear predictors
and
The hazard ratio for these two observations,
is independent of time t. Consequently, the Cox model is a proportional-hazards model.
Remarkably, even though the baseline hazard is unspecified, the Cox model can still be
estimated by the method of partial likelihood, developed by Cox (1972) in the same paper
in which he introduced the Cox model. Although the resulting estimates are not as
efficient as maximum-likelihood estimates for a correctly specified parametric hazard
regression model, not having to make arbitrary, and possibly incorrect, assumptions about
the form of the baseline hazard is a compensating virtue of Cox’s specification. Having
fit the model, it is possible to extract an estimate of the baseline hazard.
1.4 The coxph Function
The coxph function is used to fit proportional hazards regression model.
coxph(formula, data=parent.frame(), weights, subset,
na.action, init, control, method=c("efron","breslow","exact"),
singular.ok=TRUE, robust=FALSE,
model=FALSE, x=FALSE, y=TRUE,...
)
Example:
# Create a simple data set for a time-dependent model
test2 <- list(start=c(1, 2, 5, 2, 1, 7, 3, 4, 8, 8),
stop =c(2, 3, 6, 7, 8, 9, 9, 9,14,17),
event=c(1, 1, 1, 1, 1, 1, 1, 0, 0, 0),
x =c(1, 0, 0, 1, 0, 1, 1, 1, 0, 0) )
summary( coxph( Surv(start, stop, event) ~ x, test2))
Result:
Call:
coxph(formula = Surv(start, stop, event) ~ x, data = test2)
n= 10
coef exp(coef) se(coef)
z p
x -0.0211
0.98 0.795 -0.0265 0.98
exp(coef) exp(-coef) lower .95 upper .95
x
0.98
1.02 0.206
4.65
Rsquare= 0 (max possible= 0.84 )
Likelihood ratio test= 0 on 1 df, p=0.979
Wald test = 0 on 1 df, p=0.979
Score (logrank) test = 0 on 1 df, p=0.979
1.5 DNA Microarray Technology
The introduction of DNA microarray technology is a technical advance in biomedical
research. DNA microarrays rely on the hybridization properties of nucleic acids to
monitor DNA or RNA abundance on a genomic scale in different types of cells.
Hybridization refers to the annealing of two nucleic acid strands following the basepairing rules. Nucleic acid strands in a duplex can be separated, or denatured, by heating
to destroy the hydrogen bonds.
DNA microarray technology permits simultaneous measurements of expression levels for
thousands of genes, which offers the possibility of a powerful, genome-wide approach to
the genetic basis of different types of tumors. The genome-wide expression profiles can
be used for molecular classification of cancers, for studying varying levels of drug
responses in the area of pharmacogenomics and for predicting different patients' clinical
outcomes. The problem of cancer class prediction using the gene expression data, which
can be formulated as predicting binary or multi-category outcomes, has been studied
extensively and has been demonstrated great promise in recent years. However, there has
been less development in relating gene expression profiles to censored survival
phenotypes such as time to cancer recurrence or death due to cancer. Due to large
variability in time to cancer recurrence among cancer patients, studying possibly
censored survival phenotypes can be more informative than treating the phenotypes as
binary or categorical variables.
From a statistical perspective, one challenge to studying time to event outcome results
from right censoring during patient followup, since some patients may still be event-free.
These patients are termed right-censored, and for these patients, we only know that the
time to event is greater than the time of last followup. An additional challenge is in the
microarray gene expression data itself. Microarray gene expression data is often
measured with great deal of background, irrelevant readings, and the sample size of
tissues or patients is usually very small compared to the number of genes measured by
expression arrays. In addition, there is a potential of high collinearity among many genes.
Censoring of patients proves difficult when compared to binary or continuous
phenotypes. A frequent approach to relating gene expression profiles to survival
phenotypes is to first group tumor samples into several clusters based on gene expression
patterns across many genes, and then to use the Kaplan-Meier (KM) curve or the log-rank
test to indicate whether there is a difference in survival time among different tumor
groups. Another approach is to cluster genes first based on their expression across
different samples, and use the sample averages of the gene expression levels in a Cox
model for survival outcome. Both approaches suffer the drawback that the phenotype
information is completely ignored in the clustering step and therefore may
result in loss of efficiency. Additionally, results will potentially be sensitive to the
clustering algorithm and distance metrics employed, as well as the number of clusters
chosen.
Partial least squares (PLS) (Wold, 1966) is a method of constructing linear regression
equations by constructing new explanatory variables or factors or components using
linear combinations of the original variables. The methods can be effectively applied to
the settings where the number of explanatory variables is very large (Wold, 1966;
Garthwaite, 1994). Different from the principal components (PC) analysis, this method
makes use of the response variable in constructing the latent components. The method
identifes linear combinations of the original variables as predictors and uses these linear
components in the standard regression analysis. Nyuyen and Rock (2002) applied the
standard PLS methods of Wold (1966) directly to survival data and used the resulted PLS
components in the Cox model for predicting survival time. The approach did not really
generalize the linear PLS to censored survival data, but applied it directly. However, such
direct application of the Wold algorithm to survival data is questionable and indeed does
not seem reasonable since it treats both time to event and time to censoring as the same.
Park et al. (2002) reformulated the Cox model as a Poisson regression and applied the
formulation of PLS for the generalized linear models to derive the PLS components.
However, such reformulation introduces many additional nuisance parameters and when
the number of covariates is large, the algorithm may fail to converge. In addition, Park et
al. (2002) did not evaluate how well the model predicts the survival of a future patient.
A different extension of PLS to the censored survival data in the framework of the Cox
model by providing a parallel algorithm for constructing the latent components is the
Partial Cox Regression Model (PCR).
2. Partial Cox Regression Method
2.1 Introduction
The Partial Cox Regression (PCR) method involves constructing predictive components
by repeated least square fitting of residuals and Cox regression fitting. These components
can then be used in the Cox model for building a useful predictive model for survival.
The key difference from the standard principal components Cox regression analysis is
that in constructing the predictive components, the PCR method utilizes the observed
survival/censoring information.
The Partial Cox Regression method can be very useful in building a parsimonious
predictive model that can accurately predict the survival of future patients based on the
gene expression profile and survival times of previous patients.
2.2 Method for Constructing the Partial Cox Regression Model
Suppose that we have a sample size of n from which to estimate the relationship
p
between the survival time and the gene expression levels X1
T ( i 1) j ˆ ( i 1) jV( i 1) j
j1
,
… , XP of p genes. Due to censoring, for i = 1, … , n, the ith datum in the sample is
denoted by (ti , δi , xi1 , xi2 , … xip), where, δi is the censoring indicator and ti is the
survival time if δi = 1 or censoring time if δi = 0, and {xi1 , xi2 , … , xip}’ is the
vector of the gene expression level of p genes for the ith sample. Let xj = {x1j , x2j ,
…, xnj}’ be the vector of gene expression levels of the jth gene over n samples. The
aim is to build the following Cox regression model for the hazard of cancer
recurrence or death at time t
λ (t) = λo (t) exp (β1T1 + β2T2 + … + βkTk)
= λo (t) exp (f(X)),
(1)
where each component Tk and the risk score function f(X) is a linear combination of
X = {X1 , X2 , … , Xp}. In this model, λo(t) is an unspecified baseline hazard function.
Following the idea of PLS and adopting the principle that when considering the
relationship between the hazard and some specified X variable, other X variables are
not allowed to influence the relationship directly but are only allowed to influence it
through the components Tk, develop the following procedure to determine the
components sequentially. To construct the first component, first define
V1j = Xj - x . j
(2)
where x . j = 1/n∑ni=1 xij . The vector of the sample values of V1j is v1j = {v11j, ..., vn1j}
= xj - x .j and therefore the sample mean of V1j is zero. Then for each gene j, fit the
following Cox model
λ(t) = λo(t) exp(β1j V1j)
based on the sample value of V1j and obtain the maximum partial likelihood estimate
(MPLE) of β1j, denoted by βˆ1j. Then, each βˆ1jV1j provides an estimate of the log
relative hazard in the hazard function. To reconcile these estimates, set T1 equal to the
weighted average, so
p
T1 1 j ˆ1 jV1 j
(3)
j1
where ω1j is a weight with Σω1j = 1. It is easy to see that the sample mean of T1 is
also zero. Note that T1 is a special type of compound covariates advocated by Tukey
(1993) in a clinical trial setting when there are many covariates.
Note that the X variables potentially contain further useful information for predicting
the risk of recurrence or survival. So, one should not stop at the T1 step. The
information in Xj that is not in T1 may be estimated by residuals from a regression of
V1j (equivalently, Xj) on T1, and denote the residual as V2j , which can be written as
V2 j V1j -
V1j T1
T1T1
(4)
T1
Similarly, the contribution of the residual information in V1j to the variability in the
risk of recurrence or death after adjusting T1 can be estimated by performing the
following Cox regression analysis,
λ(t) = λo(t) exp(β1T1 + β2jV2j)
and denote the MPLE of β2j as βˆ2j . Then each βˆ2j V2j provides an estimate of the log
relative hazard after adjusting for T1. Define the next component T2 as
p
T2 2 j ˆ 2 jV2 j
(5)
j1
where ω2j s are the weights. Note here the sample mean of T2 is zero.
This procedure extends iteratively in a natural way to give component T2, … , TK,
where the maximum value of K is the sample size n. Specifically, suppose that
T i (i ≥ 1) has just been constructed for variables Vij , j = 1, …, p. To obtain Ti+1, first,
Vij is regressed against Ti and denote the residual as V (i+1)j , which can be written as
V(i j) j Vij -
VijTi
TiTi
Ti
Then, perform the following Cox regression analysis for each j,
(6)
λ(t) = λo(t) exp (β1T1 + β2T2 + … , βiTi + β (i+1)j V(i+1)j)
and denote the MPLE of β(i+1)j as β^ (i+1)j . Then, construct Ti+1 as
p
T(i j) (i 1) j ˆ(i 1) jV(i 1) j
(7)
j1
where ω(i+1)j s are the weights. We call the above procedure the partial Cox regression
(PCR) and the components T1, T2, … constructed the PCR components.
It is easy to verify that these components are mutually uncorrelated with sample mean
of zero. In addition, it is also easy to verify that the procedure gives precisely the PLS
in the case of linear model and when the simple linear regression is used in place of
the Cox regression. In constructing the PCR weights, let ωij var(Vij), i.e., variables
with large variance are given larger weights. This weight was also suggested in the
PLS literatures (e.g., Garthwaite, 1994). An alternative is to use equal eights of 1/p,
which aims to spread the load among the X variables in making predictions.
Here, only the variance weights are used. After the components T1, T2, … , Tk are
determined, model (1) is used for estimating the hazard function by the standard
partial likelihood method. After an estimate of the regression model (1) has been
determined, equations (3)-(7) can be used to write the risk score function f(X) in
model (1) in terms of original variables X, rather than the components T1, …, Tk , i.e.,
f(X) = Σ pj=1 β*j V1j = Σ pj=1 β*j (Xj - ‾x. j ),
for some coefficients β*j . This can then be used for estimating the hazard function for
future samples on the basis of their X values. Note that the sample mean of the score
function f(X) is zero. Finally, by examining the coefficients of X values in the final
model, one can rank the gene effects by the absolute values of the coefficients.
3. Method Implementation in R
Partial Cox regression function - PCRf
This function output the k PCR components for each individual in the
data set (comp, matrix n by k) and for each component, the
coefficients (coef, matrix m by k)
Program automatically centers and standardizes predictors.
Arguments:
x:
time:
n*m predictor matrix, with m predictors.
survival time for n observations.
surv.st: censoring status for n observations.
k:
number of output PCR components.
Program output k PCR components in comp each PCR's corresponding
coefficients output are in coef
PCRf <- function(x, time, surv.st, k)
{
x <- as.matrix(x)
nm <- dim(x)
n <- nm[1]
m <- nm[2]
ti <- matrix(0, n, k)
beta <- matrix(0, m, k)
V2 <- matrix(0, n, m)
one <- rep(1, n)
para <- matrix(0, m, k)
coef <- matrix(0, m, k)
meanx <- drop(one %*% x)/n
x <- scale(x, meanx, FALSE)
V1 <- x
space1 <- NULL
for(i in 1:k)
{
normx <- sqrt(drop(one %*% (V1^2)))
for(j in 1:m)
{
fit <- coxph(Surv(time, surv.st) ~
cbind(space1, V1[, j]))
sdf <- fit$coefficients[i]
names(sdf) <- NULL
beta[j, i] <- (sdf * normx[j])/sum(normx)
ti[, i] <- ti[, i] + beta[j, i] * V1[, j]
}
if(i == 1)
coef[, i] <- beta[, i]
else coef[, i] <- beta[, i] - coef[, 1:(i - 1)]
%*% t(t(beta[, i]) %*% para[, 1:(i - 1)])
space1 <- cbind(space1, ti[, i])
for(j in 1:m)
{
fit <- lm(V1[, j] ~ ti[, i] - 1)
V2[, j] <- fit$residuals
para[j, i] <- fit$coefficients
}
names(para) <- NULL
V1 <- V2
cat("loop=", i, "\n")
}
list(comp = ti, coef = coef)
}
4. Results
Data used is the diffuse large B-cell lymphoma (DLBCL), the most common subtype of
non-Hodgkin's lymphoma, can be found on the site, http://www.broad.mit.edu/cgibin/cancer/datasets.cgi.
The PCR components were built based on the gene expression data of the genes. The
p-values for the first ten PCR components for different number of genes are shown below
which shows that only the first 3 or 4 PCR components are strongly associated with the
survival in the univariate Cox regression analysis, indicating that the PCR components
result in dimension reduction. These components are significantly associated with the risk
of death at the 0.05 level; all other PCR components are not significant.
The p-values for univariate Cox regression analysis for the first 10 PCR components:
No.
1
2
3
4
5
6
7
8
9
10
PCR
(no. of genes = 1836)
0
3E-10
2E-02
0.233
0.223
0.280
0.829
0.824
0.552
0.889
PCR
( no. of genes = 506)
0
2E-11
2E-06
4E-05
0.066
0.233
0.199
0.516
0.514
0.994
The components which are significantly related to the risk of death at the p = 0.05 level in
univariate Cox regression analysis are used to build the final predictive model.
The mean of the risk scores of the patients in the training set, which is zero, is used as the
cutoff point to divide the patients into high and low risk groups. Based on the estimated
coefficients, the risk scores for patients in the testing data set is estimated and divided
these patients into two risk groups.
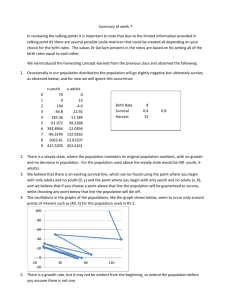
The median survival times are 10 years and 2 years respectively for the low and high-risk
groups defined by the PCR model (p = 0.0033). Following figure shows the survival
curves for two groups of patients in the testing set defined by having positive or negative
risk scores.
Figure 1. Results from analysis of DLBCL dataset
References:
Hongzhe Li, Jiang Gui (2004). Partial Cox Regression Analysis for High-Dimensional
Microarray Gene Expression Data.
Akritas MG (1994): Nearest neighbor estimation of a bivariate distribution under random
censoring. Annals of Statistics, 22:1299-1327.
Garthwaite PH (1994): An interpretation of partial least squares. Journal of the American
Statistical Association, 89, 122-127.
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh
ML, Downing JR, Caligiuri MA, Bloomfield CD, ,Lander ES: Molecular Classification
of Cancer (1999): Class Discovery and Class Prediction by Gene Expression Monitoring.
Science , 286:531-537.
Heagerty PJ, Lumley T, Pepe M (2000): Time dependent ROC curves for censored
survival data and a diagnostic marker. Biometrics, 56:337-344.
Nyuyen D, Rocke DM (2002): Partial Least Squares Proportional Hazard Regression for
Application to DNA Microarray Data. Bioinformatics, 18, 1625-1632.
Park PJ, Tian L, Kohane IS (2002): Linking Expression Data with Patient Survival Times
Using Partial Least Squares. Bioinformatics, 18: S120-127.
Rosenwald A, Wright G, Chan W, Connors JM, Campo E, Fisher R, Gascoyne RD,
Muller-Hermelink K, Smeland EB, Staut LM (2002): The Use of Molecular Proing to
Predict Survival After Themotheropy for Diffuse Large-B-Cell Lymphoma. The New
England Journal of Medicine, 346:1937-1947.
Tukey JW (1993). Tighening the clinical trial. Controled Clinical Trials, 14: 266-285.
Wold H (1966): Estimation of principal components and related models by iterative least
squares. In P.R. Krishnaiaah (Ed.). Multivariate Analysis. 391-420. New: Academic
Press.
URL
http://repositories.cdlib.org/cbmb/partialcox
www.accessexcellence.org/RC/VL/GG/microArray.html