PART I
advertisement

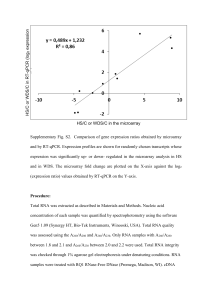
3.3.2.2.3.6 Amplification of differentially expressed genes Two techniques-differential screening and subtraction hybridization - can be used to isolate gene transcripts that are present at different levels in two RNA populations. However, these two approaches require large amounts of RNA to synthesize sufficient quantities of an enriched cDNA probe for library screening. In contrast, RT-PCR can be used to generate cDNAs from very small amounts of mRNA. Here, two basic RT- PCR cloning approaches have been described to identify differentially expressed genes. 1 Figure 8. Competative RT-PCR uses a mimic cRNA to quantify the amount of target mRNA in an experimental sample based on a ratio of PCR products. a) Differential display reverse transcriptase PCR (DDRT-PCR): This technique, developed by Liang and Pardee (1992), is based on the principle of two-dimensional gel electrophoresis as a rapid screening procedure for differrential display of proteins that were metabolically labeled in vivo or in vitro. The appearance or disappearance of 2 protein spots in the two-dimensional array could provide information about cellular changes in protein synthesis. Liang and Pardee applied the same differential display approach to mRNA by developing a method to array RTPCR products on standard DNA sequencing gels. DDRT-PCR provides the potential to clone differential RT-PCR cDNA products by physically excising radiolabeled bands from an acrylamide gel. Modified oligo dT primers are used for reverse transcription that anneal to a subset of poly A+ mRNAs owing to differences in dinucleotides at the 3' end of the primer. Figure.10 shows how a 5' – dT-GC - 3' primer is used to synthesize cDNA from a subset of RNAs containing the appropriate 3' dinucleotide adjacent to the poly A tail.The product is then PCR amplified in the presence of 33P-dATP and a known primer. Of these first-strand cDNAs, only a fraction serve as appropriate templates for separate PCR reactions containing a known arbitrary primer. Candidate RT-PCR products are excised from the gel, reamplified with the same primer pair, and used as probes on conventional Northern blots to verify that they correspond to differentially expressed gene transcripts. The false positive rate with DDRT-PCR can be highly variable and therefore it can be best used as a screening procedure rather than a cloning strategy. A similar RT-PCR screening technique called RAP-PCR (RNA arbitrarily primed PCR), is based on a genomic DNA fingerprinting strategy. The primary difference between these two methods is that RAP-PCR uses the same random primer for both reverse transcription and PCR amplification steps, which eliminates bias toward the noncoding 3' poly A+ sequences. b) Suppression subtraction hybridization (SSH): It was developed a PCR-based strategy to clone differentially expressed gene transcripts. Although it is technically not an RT-PCR approach, it does require the synthesis of double-stranded DNA as a starting point for exponential PCR amplification of gene transcripts present at higher concentration in one of the two mRNA pools. The basis for differential amplification in SSH is two-fold. First, conditions are used that promote rapid reassociation kinetics with excess "driver" cDNA sequences to normalize two "tester" cDNA pools. Doublestranded cDNA is made from mRNA representing tester (containing gene transcripts of interest) and driver (lacks appreciable amounts of desired gene transcript) transcripts (Fig. 11). The tester cDNA is first digested with the base cutting restriction enzyme (Rsal) to create bluntended termini and 3 Figure 10. Schematic diagram of DDRT-PCR showing how a 5’ – dT- GC- 3’ primer is used to synthesizes cDNA from a subset of RNAs containing the appropriate 3’ dinucleotide adjacent to the poly A tail. 4 Figure 11. The flow diagram of suppression subtraction hybridization technique. then divided into two equal portions, and distinct primers (Adp 1 and Adp 2) are ligated to the two cDNA pools. In the next step, separate rapid reassociation hybridization reactions are performed (rxn 1a and rxn 1b) with each tester cDNA pool and a mass 5 excess of driver cDNA. Following this driver testei hybridization reaction, the two tester pools are mixed without prior denaturation and the unhybridized single-strand low abundance molecules are allowed to anneal under conditions to initiate a second hybridizatior reaction (rxn 2). In the subsequent PCR amplification step, a pair of adapter primers exponentially amplifies these unique ‘tester’ duplexes. Second, the PCR amplification of nondifferential cDNAs is suppressed because undesirable sequences contain inappropriate priming sites or are capable of forming intrastrand "panhandle" structures that are poor substrates for PCR. The combined PCR effect of exponential amplification of selective tester duplex cDNAs, with the resulting suppression of common sequence cDNAs, can potentially result in a 102 - to 103 - fold enrichment of differentially expressed tester cDNA sequences. The amplifier cDNAs are used to make an enriched plasmid library, which can be randomly sequenced or screened with a differential cDNA probe. 3.3.2.2.3.7 Exhaustive limiting dilution PCR method This method is based on: (1) optimization of the PCR so that amplification of an endogenous control gene will take place in an all-or-nothing fashion, derived from the terminal plateau phase of the PCR (2) the premise that one or more targets in the reaction mixture (e.g. GMO) will give a positive result. Accurate quantitation is achieved by performing multiple replicates at serial dilutions of the material(s) to be assayed. At the limit of dilution, where some end points are positive and some are negative, the number of targets present can be calculated using Poisson statistics from the proportion of negative end points. An advantage of this method is that coamplification of added reporter DNA is not required. However, caution should be exercised because of potential contamination of PCR reactions owing to various dilutions and manipulations. 6