Genetics & Molecular Biology - American Academy of Optometry
advertisement

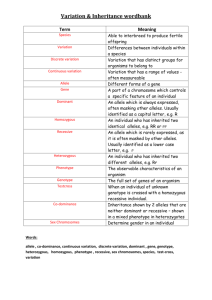
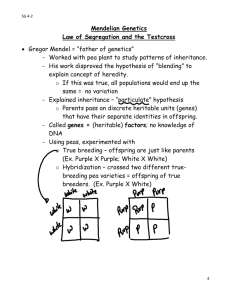
I. Inheritance patterns A. Introduction. 1. Because the role that genetic factors play in pathologic processes is now better understood, the importance of genetics in optometry and medicine has increased. Scientific advances have made it possible to identify genetic diseases more precisely, to provide better genetic counseling and more accurate prenatal diagnosis for genetic diseases, and to improve the health of many people affected with such conditions. 2. Until molecular genetic testing becomes more common, the genetic test that all clinicians can administer is the taking of the family history. For selected patients this would involve the drawing of at least a three-generation pedigree. This would facilitate the discovery of the inheritance pattern of the condition and could aid in the diagnosis of the condition. B. Major types of genetic disease 1. Chromosome abnormalities are deviations from the normal chromosome number or structure. Examples include Down syndrome and Turner syndrome. 2. Single gene (Mendelian or monogenic) disorders are conditions that are produced by the effects of one gene or a gene pair. Such traits are usually transmitted in simple patterns as originally described by Gregor Mendel. a. Autosomal dominant traits are transmitted on the autosomes (i.e., chromosomes other than the X or Y) and are expressed when only a single copy of the mutant gene is present. Tritan (blue cone pigment) color deficiency is an example of an autosomal dominant disorder. b. Autosomal recessive traits are transmitted on the autosomes and are only expressed when both copies of a gene are mutant. Tay-Sachs disease is an autosomal recessive disorder. c. X-linked traits are transmitted on the X chromosome. An example of an X-linked disorder is protan (red cone pigment) color deficiency. 3. Multifactorial traits result from the combined effects of multiple genetic and non-genetic influences. Examples include common kinds of diabetes mellitus and glaucoma. C. The incidence of genetic disease and other congenital anomalies apparent by 25 years of age is about 79/1000 livebirths (7.9%). This does not include most of the common diseases of adulthood such as hypertension, Type 2 diabetes mellitus, coronary artery disease, and cancer, which have a multifactorial etiology but have an age of onset usually older than 25 years. D. The prevalence of monogenic disorders. 1. Autosomal dominant: > 50% 2. Autosomal recessive: 33% 3. X-linked: 10% II. Chromosome anomalies. A. Introduction. 1. Euploidy means that the chromosome number per cell is an integral multiple of the haploid number, N = 23. 2. Diploidy indicates the normal condition where a human cell has 2N = 46 chromosomes. Normal women have a karyotype formula of 46, XX, and normal men have a karyotype of 46, XY. B. Types of chromosome anomalies. Chromosome anomalies can involve abnormal numbers of chromosomes and/or aberrations in their structure 1. Numerical anomalies can result in either aneuploidy or polyploidy. a. Aneuploidy indicates that the chromosome number per cell is not an integral multiple of the haploid number. Thus, it describes an addition or loss of one or, rarely, two chromosomes. For example: 1) Trisomy (2N+1) refers to three copies of one chromosome. 2) Monosomy (2N-1) refers to the presence of only a single copy of an autosome. b. Polyploidy refers to multiples of the haploid number where there is an addition of complete haploid sets of chromosomes (e.g., triploidy, 3N = 69). 2. Structural anomalies are rearrangements of genetic material within or between chromosomes. These may be either genetically balanced, in which there is no change in the amount of essential genetic material, or unbalanced, in which there is a gain or loss of essential chromosome segments. They include aberrations like deletions, duplications, and translocations. B. Clinical example: Down syndrome, trisomy 21 (karyotype of 47, XX, +21 or 47, XY, +21). Approximately 1 in 680 newborns has Down syndrome. The frequency is doubled at 10 to 12 weeks of pregnancy; trisomy 21 is a common finding in spontaneously aborted fetuses. 3. Infancy. The infant has a characteristic face with a flat nasal bridge, epicanthal folds, Brushfield spots (mottled or speckled spots on the iris), protruding tongue, small ears, and a flat occiput. 4. Childhood and adulthood. All affected children have mental retardation, which is usually moderate. Other features include short stature, autoimmune abnormalities, and hearing loss. 5. Life expectancy. For some individuals it is less than 50 years due to heart defects. III. Single gene alterations A. Single gene (Mendelian) inheritance describes those conditions in which single mutant genes have a significant effect on human health and in which simple patterns of inheritance are seen. Genes are recognized by the physical characteristics or traits that they determine. Pedigree analysis is essential in determining the mode of inheritance of many traits. 1. Definitions. The mode of inheritance for single gene conditions depends on whether the specific phenotype is seen in the heterozygote (dominant) or only in the homozygote (recessive). a. Gene. This term describes the linearly ordered, information-containing, hereditary factor that interacts with the environment to determine a trait. b. Alleles are alternative forms of a gene at a given locus. There may be multiple normal and abnormal alleles of any particular gene. The most common allele at a locus is labeled the wild-type or normal allele. Because human chromosomes are paired, humans have two alleles at each locus. c. A locus is the specific physical location of a gene on a chromosome. Since human chromosomes are paired, individuals have two alleles at each locus (the exceptions are genes on the Y chromosome and the X chromosome in males). d. Genotype describes the genetic constitution of an individual, which is the specific allelic makeup of an individual. e. Phenotype describes the end result of both the genetic and environmental factors giving the clinical picture or the observed expression of the gene. This can be a grossly observable characteristic of the individual or a specific protein. Genotype-phenotype correlation studies seek to define whether certain clinical features of a condition are seen with a specific mutation of the gene. Pleiotropy describes the situation where a single allele expresses several different phenotype features. f. If the alleles at a single locus are identical, the individual is said to be homozygous. The alleles can be either normal or abnormal. g. If the alleles at a single locus are different, the person is said to be heterozygous. The term usually refers to having one normal and one abnormal or mutant allele. Such heterozygous individuals are also referred to as carriers. h. A dominant condition is seen in both the heterozygote and the homozygote. This implies that a single copy of the mutant allele is enough for the condition to be expressed. These individuals usually carry a mutant allele on one chromosome and a normal allele on the homologous chromosome. There are at least four different situations where one normal allele is not enough to prevent disease: (1) Normal function may require more than 50% of normal protein or gene product to prevent disease. When loss of half of the normal protein causes disease, the situation is referred to as haploinsufficiency. This has been shown to occur with mutations is genes encoding structural proteins, certain transcription factors, and cell surface receptors. (2) Instead of deficiency or dysfunction of the protein, an abnormal protein may cause an abnormal phenotype by interfering with the function of the product of the normal allele (dominant negative effect). For example, this occurs with collagen mutations in osteogenesis imperfecta (“brittle-bone disease”). (3) On the other hand, the mutant protein may be enhanced in one or more of its normal properties through mutation (simple gain of function). For example, this occurs in the dwarfing condition achondroplasia. Or the mutant protein could become toxic to the cell by acquiring a novel property, as in Huntington disease. (4) With certain autosomal genes, an inherited dysfunction of one copy of the gene may result in pedigrees with dominantly inherited cancers (e.g., hereditary retinoblastoma). The predisposition to cancer is inherited as a dominant trait, but the mutations that precipitate cancer are recessive at the cellular level since both copies of the gene must be mutated for the cancer to develop. Thus, a random mutation can occur that renders the normal allele dysfunctional, and thus with both alleles mutated, the cell becomes cancerous. i. A recessive condition is seen only in the homozygote, which means that the mutant allele must be present on both chromosomes. Dominant and recessive refer to the expression of the clinical conditions, not to the genes themselves. j. Autosomal refers to the autosomes, which are the non-sex chromosomes. An autosomal condition results from mutant alleles on an autosome. k. X- or Y-linked refers to genes having loci on either the X or Y chromosome. With X-linked alleles, both recessive and dominant inheritance may be seen. l. A compound heterozygote is an individual with two different mutant alleles at one locus. m. A double heterozygote is an individual with two mutant alleles, each at a different locus. 2. Pedigree symbols and construction. The pattern of inheritance of many single gene traits has been deduced from examination of the family history. When the history is taken, the pedigree is a shorthand or graphic representation of the details. Individuals are characterized according to their sex, their generation, and their biologic relationship to each other. 3. Punnett square. The Punnett square is a method for showing the gametes produced by mating individuals and their possible combinations at fertilization. The normal allele is shown as a lower case letter and the mutant allele as a capital letter. The maternal gametes are shown on the left side of the square, and the paternal gametes are shown at the top of the square. The four cells of the square show the different combinations possible. Each cell represents a 25% probability of occurrence. For example, if the mother is homozygous for the normal allele and the father is heterozygous, then the offspring have a 50% chance of being heterozygous and a 50% chance of being homozygous normals. Paternal gametes Maternal gametes A a a Aa aa a Aa aa B. Autosomal dominant inheritance. More than half of the currently described traits are inherited in a dominant fashion: approximately one-third as recessive and one-tenth as X-linked. Dominant implies that the disease allele need be present only in a single copy (as in a heterozygote) to result in the phenotype. 1. The criteria of autosomal dominant inheritance include the following: a. The transmission of the trait is vertical, from generation to generation without skipping. In a typical dominant pedigree, there can be many affected members in each generation. b. Except for a new mutation or non-penetrance, every affected child will have an affected parent. Direct transmission through three generations is essentially diagnostic of dominant inheritance. c. In the mating of an affected heterozygote to a normal homozygote (the usual situation), each child has a 50% chance to inherit the abnormal allele and be affected and a 50% chance to inherit the normal allele. d. The two sexes are affected in equal numbers. Since the loci are on the autosomes (i.e., nonsex chromosomes), a sex differential is not seen unless the mutant allele involves a sex-limited structure such as the uterus. e. Father-to-son transmission is seen. f. The trait is expressed in the heterozygote but is more severe in the homozygote. 2. Features. To provide the most accurate counseling, a number of potential complicating factors are important to consider in autosomal dominant inheritance. Each of the following may alter the idealized dominant pedigree. a. New mutations. An isolated case of a dominant disorder may be the result of a new mutation, but other causes (e.g., genetic heterogeneity or a minimally affected parent) and nonpaternity should also be considered. b. Reduced penetrance. Penetrance refers to expression of the mutant phenotype and is an all-ornothing phenomenon. It is expressed as the percentage of individuals who have the mutant allele and are actually affected. If the frequency of expression of a genotype is less than 100%, reduced penetrance exists. Nonpenetrance refers to the situation in which the mutant allele is inherited but not expressed. Penetrance is a clinical term and may be influenced by factors including age (e.g., Huntington disease) or better detection methods (e.g., magnetic resonance imaging for signs of tuberous sclerosis). c. Variable expressivity. Expressivity refers to how or to what degree a particular allele is expressed in an individual. (1) Variable expressivity is common in dominant disorders, which means that all family members should be examined carefully, with the features of the condition kept in mind. (2) Affected individuals who are capable of reproducing are generally less severely affected. More severe expression may be seen in their children (i.e., they may express the full spectrum of the syndrome). d. Variation in age of onset. Before the advent of molecular testing, the diagnosis could not be reliably ruled out in a young person. e. Mosaicism. If a parent is mosaic for a disease allele because of a postzygotic mutation, he or she may appear clinically unaffected. f. Genetic heterogeneity. Different mutations, at the same locus or at different loci, may result in a similar clinical picture. In addition, the conditions may be inherited in different manners (e.g., retinitis pigmentosa may be inherited as an autosomal dominant, autosomal recessive, or Xlinked trait). 3. Clinical examples a. Marfan syndrome. (1) This is a disorder of connective tissue with variable expressivity involving the skeletal system (e.g., abnormal proportions, scoliosis, arachnodactyly), cardiovascular system (e.g., mitral and atrial valve prolapse, aortic dilatation leading to aortic dissection), and ocular system (e.g., lens dislocation, monocular diplopia, blue sclera). (2) There is an increased risk of cataract, glaucoma, and retinal detachment. (3) The diagnosis is made on clinical grounds, with involvement of at least two systems. (4) The molecular basis of Marfan syndrome is mutation of a structural gene (FBN1: fibrillin) on chromosome 15q. b. Juvenile-onset, open angle glaucoma. (1) This disorder is caused by a gene (MYOC; GLC1A) on chromosome 1. This gene codes for a protein found in the trabecular meshwork (TM) called trabecular meshwork-induced glucocorticoid response protein (TIGR). The reason for the name is that exposure of the patient to steroid induces the making of this protein in the TM (and ciliary body). The protein is also known as myocilin, and this is the term recently adopted by the Human Genome Organization. (2) This discovery allows the identification of individuals at risk for the development of the disease before vision is lost. (1) This finding also provides insight into the molecular cause of a subset of glaucoma and may lead to the development of more effective treatments. (2) The anti-inflammatory drug, diclofenac, reduces the activity of the GLC1A gene. A reformulated version of the drug is in the early-stage of human tests for the treatment of glaucoma. Table 1. Examples of AD disorders. Disorder Hereditary non-polyposis colorectal cancer Dominant otosclerosis (progressive deafness due to bony overgrowth in inner ear Familial hypercholesterolemia Neurofibromatosis Marfan syndrome Osteogenesis imperfecta Myotonic dystrophy Huntington disease Occurrence 1/200 1/100 to 2/1000 1/500 1/3,500 1/15,000 1/15,000 1/7,000 to 1/20,000 Caucasians 1/20,000 Caucasians C. Autosomal recessive inheritance. The phenotype is usually only in the homozygote, and the typical pedigree demonstrates affected male and female siblings with normal parents and offspring. Recessive inheritance is suspected when parents are consanguineous; it is considered proven when corresponding enzyme levels are low or absent in affected individuals and are at half-normal values in both parents. 1. Criteria a. The mutant gene usually does not cause clinical disease in a heterozygote. b. Individuals who inherit both mutant genes will express the disorder. c. The characteristic pattern of inheritance is horizontal. In other words, the disorder is found in a single group of brothers and sisters. The disorder is not found in multiple generations. If the trait is rare, parents and relatives other than siblings are usually clinically normal but heterozygous for the mutant allele. Thus, the parents are said to be carriers of the allele. d. In the mating of two normal heterozygotes, the segregation frequency with each pregnancy is 25% homozygous normal, 50% heterozygous normal, and 25% homozygous affected. e. If the recessive genes are alleles, all children of two affected parents are affected. f. Both sexes are affected in equal numbers. g. Parents of an affected individual may be genetically related (consanguinity). If the trait is rare, the likelihood of parental consanguinity is increased. 2. Features. A number of features are important to keep in mind before counseling families regarding autosomal recessive inheritance. a. Heterozygote frequency. Affected individuals are almost always the offspring of normal heterozygotes (not homozygotes). b. Carrier state. Carriers are assumed to be unaffected, but some may show half-normal enzyme levels (e.g., the level of hexosaminidase A in Tay-Sachs disease heterozygotes) or minimal clinical features. The majority of carriers are clinically normal, however. c. Consanguinity. In the classical description of the rare autosomal recessive trait alkaptonuria, 60% of affected children had parents who were cousins. Consanguineous individuals have a proportion of their genes in common by descent. d. Autosomal recessive phenotypes in the local population. It is important to know an area’s population when counseling because there may be particular racial groups in which a disease allele is carried more frequently because of the founder effect or heterozygote advantage. Knowledge of the local population allows appropriate screening and counseling. Examples of this include: (1) Sickle cell anemia in blacks (2) Tay-Sachs in Ashkenazi Jews (3) Cystic fibrosis in northern European Caucasians e. Heterogeneity. Clinically similar individuals may have genetic causes for their condition but have mutant alleles at different loci. For example, albinism may be recessively inherited but have different loci involved in different families. f. Most recessive genetic disorders involve an enzyme deficiency. 3. Clinical examples a. Tay-Sachs disease (1) This autosomal recessive disease is rare in the general population (1:300,000), but it has a high incidence among Ashkenazi Jews (1:3600 births). About 3% of Ashkenazi Jews are carriers. (2) The deficient enzyme is hexosaminidase A. Deficiency of the enzyme results in the accumulation of the ganglioside GM2 in the ganglion cells of the retina and brain, causing demyelination. (3) The key clinical sign is a cherry-red spot in the macula due to the accumulation of gangliosides in the retinal ganglion cells. This produces a pale fundus, with a normal reddish fovea (no ganglion cells), seen as early as 2 months. Severe visual impairment and blindness occur by 18 months. (4) Infants develop normally until 3 to 7 months. Mental retardation, deafness, dementia, spasticity, nystagmus, and ophthalmoplegia occur due to accumulation of ganglioside in the gray matter. Death occurs by 2-4 years old. (5) Widespread testing for carriers has virtually eliminated the disease in Ashkenazi Jews over the past 30 years. b. Stargardt disease / fundus flavimaculatus (1) This recessive macular dystrophy is one of the most frequent causes of macular degeneration in childhood. The onset is in the first to second decade with rapid progression, usually to 20/200 visual acuity. (2) The condition is due to a mutation in the ATP-binding cassette transporter, retina-specific (ABCR) gene on chromosome 1. The ABCR gene is now called ABCA4 and is a member of the ABC transporter superfamily which includes genes whose products are transmembrane proteins involved in energy-dependent transport of a wide spectrum of substrates across membranes (e.g., lipids, hydrophobic drugs, and peptides). The gene causing cystic fibrosis is also a member of this superfamily. (3) The protein made by the gene is called rim protein (RmP) and is found in the hairpin loop or rim of the rod photoreceptor disk membrane. The function of ABCA4 rim protein is to translocate N-retinylidine-phosphatidylethanolamine (N-retinylidine-PE), a vitamin A derivative, across the disk membrane from the disk lumen to the cytoplasm. Nretinylidine-PE forms after the conversion of 11-cis-retinal to the all-trans isoform as a result of the photopigment rhodopsin absorbing a photon. Without a functional ABCA4 gene, Nretinylidine-PE accumulates in the photoreceptor outer segment disks and forms N-retinylidineN-retinylethanolamine (A2-E), the major component of lipofuscin. Subsequently, abnormally high levels of lipofuscin buildup in the retinal pigment epithelial (RPE) cells. This may trigger RPE-cell death with the secondary degeneration of the photoreceptors. (4) Over 400 mutations have been found in ABCA4 with the majority representing missense amino acid substitutions in conserved functional domains. (5) The ophthalmoscopic findings include: loss of foveal reflex, granular appearance, beaten bronze appearance of the fovea. Some patients have a bullseye maculopathy. Yellow flecks at the level of the retinal pigment epithelium may surround the macular lesion and extend to the periphery. c. Usher syndrome (USH). (1) USH is a group of clinically and genetically heterogeneous autosomal recessive disorders. They are characterized by the combination of congenital or early-onset sensorineural deafness and retinitis pigmentosa. (2) USH is the most frequent cause of deafness accompanied by blindness. USH accounts for more than 50% of deaf-blind patients, about 18% of patients with retinitis pigmentosa, and 3-6% of the congenital deaf patients. The prevalence of USH is between 1/16,000 and 1/50,000. As with other recessive conditions, certain population isolates (e.g., a FrenchAcadian group in Louisiana) show a high prevalence. (3) At least 12 different chromosomal loci have been found that cause USH, and 6 genes have been identified. This suggests that many proteins are important in the maintenance of normal retinal, auditory, and vestibular function. (4) Three clinical subtypes have been defined: USH1, USH2, and USH3. Table 2. Examples of AR disorders. Disorder Occurrence Sickle cell disease 1/400 Blacks Cystic fibrosis 1/2,000 to 1/4,000 Caucasians Stargardt disease 1/8,000 to 1/10,000 (approximate) Phenylketonuria 1/20,000 Caucasians Tay-Sachs 1/3,000 Ashkenazi Jews Thalassemia 1/50 to 1/100 Southeast Asians/Mediterraneans Usher syndrome 1/16,000 to 1/50,000 Oculocutaneous albinism 1/35,000 D. X-linked inheritance can be either recessive or dominant, although in patients with a recessive X-linked condition, the phenotype is usually only observed in the male. X-linked inheritance is suspected when several male relatives in the female line of a family are affected. Because males have only one X chromosome, they are hemizygous, not heterozygous, for X-linked genes. 1. Criteria a. X-linked disorders are due to mutant alleles on the X chromosome. Since males have only one X chromosome, X-linked mutant genes are fully expressed in males. In females heterozygous for an X-linked mutant gene, an X-linked trait may or may not be expressed because of lyonization. If a disease is rarely expressed clinically in a heterozygous female, it is called X-linked recessive. In fact, most cases of X-linked inheritance are recessive. b. If the trait is rare, parents and relatives (except maternal uncles and other male relatives in the female line) are usually normal. c. Hemizygous affected males have neither affected sons nor daughters. All daughters of affected males are obligate heterozygotes (i.e., they do not show a mutation clinically but carry the mutant allele). There is no male-to-male transmission. d. Female heterozygous carriers are clinically normal but will transmit the trait to sons (i.e., hemizygous affected) 50% of the time. Daughters of carrier mothers are normal heterozygotes 50% of the time and normal homozygotes 50% of the time. e. Most often, affected daughters are produced only by matings of heterozygous females with affected males. f. Except for those with new mutations, every affected male is born of a heterozygous female. g. If the trait is dominant, all female offspring of affected males will themselves be affected. On average, 50% of either male or female offspring of an affected female heterozygote will be affected. Unlike autosomal dominant pedigrees, there is no male-to-male transmission. 2. Features. Some specific features of X-linked inheritance that cause problems are important to keep in mind. a. The sporadic case. If a male is affected but there are no other affected family members, the question is whether the condition is the result of a new mutation or whether the mother is heterozygous. Heterozygous females may be detected by subtle clinical features, intermediate enzyme levels, or molecular methods. In about 2/3 of cases of X-linked recessive diseases that are lethal in childhood and in which there is only one affected male in the family, the mother is heterozygous. In 1/3 of cases, the son has a new mutation. b. Heterogeneity. Diseases with similar clinical features may be inherited through differing mechanisms. (1) Albinism is usually inherited as an autosomal recessive condition, with involvement of eyes, hair, and skin. (2) An X-linked recessive form, ocular albinism, primarily involves the eyes, with little skin and hair involvement. The condition may be misdiagnosed in a blond child and incorrect counseling provided. (3) A good clinical examination and family history are essential. c. Affected females. X-linked conditions affecting females can be explained by a number of mechanisms, including random X chromosome inactivation (lyonization). 3. Clinical examples. The genes encoding the red and green photopigments are arranged in a head-totail tandem array on the X-chromosome (Xq28). These arrays are made up of a single red photopigment gene and one or more green photopigment genes. About 25% of male Caucasians have one green photopigment gene, about 50% have two, and the rest have three or more. Only the first two genes of an array are expressed in the retina enough to influence the color vision phenotype. The red and green photopigment proteins are very similar. They differ in amino acid sequence at only 15 positions. This similarity and closeness of the genes on the X-chromosome lead to frequent adverse recombination events and thus account for a relatively high frequency of red/green color deficiency. a. Protan (red cone pigment) color deficiency. About 2% of western European males have a color deficiency due to red cone pigment involvement. b. Deutan (green cone pigment) color deficiency. About 6% of western European males have a color deficiency due to green cone pigment involvement. Table 3. Examples of X-linked recessive disorders. Disorder Occurrence G6PD deficiency 0-65/100 (geographically very variable) Red/green color deficiency 8/100 Caucasian males 4-5/100 Asian males 1-4/100 Black and native American males Fragile X syndrome 1/1,000 to 1,500 males 1/2,500 females Duchenne Muscular Dystrophy 1/3,500 males Hemophilia A 1/5,000 males E. Mitochondrial inheritance. Human cells have hundreds of mitochondria dispersed throughout the cytoplasm, each containing a number of circular DNA molecules. Mutations involving the mitochondrial DNA are now known to account for a small number of genetic conditions. 1. Criteria. a. Each mitochondrion contains a number of copies of the circular genome. Mitochondrial enzymes are usually encoded by nuclear genes, but some of the cytochrome oxidase enzymes are derived solely from mitochondria. b. Almost all mitochondrial DNA is maternally inherited. Pedigrees with mitochondrial inheritance should show that all children of an affected mother are affected and all children of an affected father are normal. c. Specific tissues have different proportions of mitochondria. Rich areas include striated and cardiac muscle, the kidney, and the central nervous system (CNS), including the eyes. Therefore, mitochondrial diseases are commonly myopathies, neurologic syndromes, cardiomyopathies, and optic neuropathies. 2. Clinical examples 1. Kearns-Sayre syndrome is a sporadic condition with onset before 20 years of age. Characteristics include progressive external ophthalmoplegia (ptosis with paralysis of extraocular muscles), retinal pigment abnormalities, heart block, and cerebellar ataxia. Studied patients exhibit deletions of muscle-derived mitochondrial DNA. 2. Leber hereditary optic neuropathy (LHON). LHON is a rare, maternally inherited, neurodegenerative disease that is characterized by acute or subacute, painless, severe, sequential bilateral loss of central vision and, ultimately, optic atrophy in healthy young persons. IV. Multifactorial (complex) disorders A. Multifactorial (complex) disorders are the most common type of disorders seen in practice. B. Multifactorial inheritance is a pattern of inheritance that results from the interaction of one or more genes with environmental factors. Thus, a multifactorial trait has a familial nature and an environmental dependence. 1. Polygenic traits are traits generated by the combined action of many genes. 2. If environmental factors are also involved then the traits are multifactorial. These traits are also called complex traits. C. Most normal phenotypic differences among individuals are due to multifactorial inheritance. This includes differences in height, hair and skin colors, and intelligence. D. Clinical characteristics of complex disorders. 3. These disorders can be common (> 1/5000 births). 4. The disorder tends to be familial, but there is no distinctive pattern of inheritance within a single family. 5. The risk of first-degree relatives is approximately the square root of the population risk. Thus, the lower the population incidence, the greater the relative increase in risk for first-degree relatives. 6. Recurrence risk is proportional to the number of family members who already have the disease. This is due to the fact that the involvement of a number of family members indicates a high concentration of adverse alleles. 7. Recurrence risk is proportional to the severity of the condition in the proband. This is due to the fact that both recurrence risk and severity depend on the concentration of adverse alleles. 8. The recurrence risk is the same for all relatives who share the same proportion of genes. For example, each sibling, child, and parent shares half of his or her genes on the average. The recurrence risk in such first-degree relatives would be expected to be about the same. 9. The frequency of disease in second degree relatives is much lower than in first degree relatives. However, the frequency declines less rapidly for more distant relatives. 10. The concordance rate in monozygotic (MZ) twins is significantly greater than in dizygotic (DZ) twins. a. MZ or identical twins originate from a single zygote that divides into two embryos during the first two weeks of development. Their DNA is identical. DZ or fraternal twins come from two sperm and two eggs. Thus, they are like ordinary siblings who share half of their alleles. b. If both twins have a particular disease or condition, they are said to be concordant. If only one has the disease, they are discordant. 11. Multifactorial diseases are more common among the children of consanguineous parents. This is due to the fact that parents who are related are more likely to share similar disease-predisposing genes. 12. Complex diseases tend to occur more frequently in one sex than in the other. For example, systemic lupus erythematosus (SLE) is more common in females, but pyloric stenosis is more common in males. 13. Recurrence risk is higher for relatives of the less susceptible sex. This is because a higher concentration of harmful alleles is necessary for disease expression in the less susceptible sex. E. Clinical examples. 1. Type 1 diabetes mellitus (T1DM) accounts for 5-10% of the individuals with diabetes mellitus. Familial patterns and twin studies indicate that there is a genetic basis. Two loci, IDDM1 (in the major histocompatibility complex on chromosome 6p21.3) and IDDM2 (adjacent to the 5-prime end of the human insulin gene on chromosome 11p15.5) account for 40-50% of the genetic predisposition. At least 17 loci have been found. 2. Type 2 diabetes mellitus (T2DM) is about 10 times more prevalent than T1DM and accounts for 9095% of patients with diabetes. And about 6% of the population in the United States has diabetes. Twin studies, family studies, and linkage studies indicate a strong genetic basis. At least 7 single gene defects are known to cause T2DM, but these account for 5% or less of all T2DM. 3. Age-related macular degeneration (AMD). a. A SNP in the complement factor H gene (CFH) on chromosome 1 accounts for 20-50% of the overall risk of developing AMD. If the patient carries a single copy of the mutant allele, there is a 2- to 4-fold increased risk of AMD. If the patient is carrying two copies of the mutant allele, there is a 3- to 7-fold increase in the risk of AMD. In case-control studies it has been found that the CFH allele (either heterozygous or homozygous) accounts for an odds ratio (OR) of 5.3 and a significant population attributable risk (AR) as high as 68%. This is consistent with AMD damage being a result of chronic inflammation, mediated by the complement system. CFH modulates the complement cascade by inactivating components of the cascade and by binding initiation factors such as C-reactive protein. It may be that the change due to the SNP affects the binding sites for heparin and C-reactive protein, thus altering CFH’s ability to suppress complement-related damage to arterial walls and ultimately leading to vessel injury and subsequent neovascular AMD. Hence, atrophic tissue damage and neovascularization are potential consequences of chronic inflammation, mediated by the complement system. In addition, the known AMD risk factors (age, smoking, and diet) correlate with complement activity, and complement proteins have been found in drusen. Alternatively, this SNP may have a direct role in the formation of soft drusen. b. Similarly, a SNP in the LOC387715 gene on chromosome 10 accounts for as high as 57% of the overall risk of developing AMD. If the patient carries a single copy of the mutant allele, there is about a 3-fold increased risk of AMD. If the patient is carrying two copies of the mutant allele, there is a 5- to 8-fold increase in the risk of AMD. In case-control studies it has been found that the LOC387715 allele (either heterozygous or homozygous) accounts for an OR of 5.0 and an AR as high as 57%. c. There appears to be an independent contribution of the two gene loci to disease risk. Homozygosity for risk alleles at both loci CFH and LOC387715 is associated with a very high OR of 57.6 which applies to about one in 200 individuals. The next lower category of risk (ORs between 16 and 20) could affect as many as one in 23 individuals. These considerations could make predictive DNA testing a tempting option. However, the knowledge of being a carrier of risk alleles is currently not matched by adequate options for preventive strategies or possible treatment modalities. Table 4. Examples of multifactorial (complex) disorders. Categories Examples Ocular Primary open-angle glaucoma, AMD, myopia Birth defects Cleft palate/lip, spina bifida, anencephaly Cancer Bowel, breast, colon, melanoma, etc. Cardiovascular Hypertension, heart disease, hypercholesterolemia Metabolic Diabetes Mental illness Schizophrenia, bipolar disorder Muscular/skeletal Arthritis, rheumatic disorders, osteoporosis Respiratory Asthma, allergies, emphysema Skin disorders Psoriasis, nevi, eczema V. Pedigree Drawing Exercise. A. Learn to use family history (FH) as a screening tool. 1. All clinicians take family histories. 2. Because of the genomics revolution, the purpose and scope of taking a FH have changed. 3. Increased understanding of the role of genetics in health means that you need to expand your FH to include information on genetic factors. B. Risk factors for disease 1. Genes, environment, and behaviors interact with each other to cause disease. 2. Family history helps capture the effects of these interactions on disease risk. C. An efficient , accurate family history/pedigree can be used to: D. E. F. G. H. I. J. 1. develop a differential diagnosis 2. make a diagnosis 3. distinguish genetic from other risk factors 4. identify patterns of inheritance 5. identify medical risks for other relatives 6. calculate risks 7. decide an approach to genetic testing (e.g., whom to test, which type of test to order) 8. make decisions on management, prevention, and surveillance 9. assess reproductive options 10. develop patient rapport and trust 11. see family dynamics (e.g., sources of conflict and support) 12. educate patients and families; clarify misconceptions In select cases, the FH information should be converted into a diagram of the biological relationships of your patient with his or her family members – a pedigree. A pedigree is indicated where: 1. The patient presents with a concern about an inherited disease. 2. The patient presents for a routine screening. The patient could fill out a FH questionnaire. 3. The patient presents with an established diagnosis of a disease that possibly has a genetic basis. 4. The patient indicates a significant FH of an ocular disease or a systemic disease with an ocular component. Learn to take a minimum of a three-generation pedigree from selected patients. Maintain and update the pedigree in these patients. Use FH to guide and inform prevention (see handout). Family History Tools 1. U.S. Surgeon General’s Family History Initiative (http://www.hhs.gov/familyhistory/) goals: a. increase the public’s awareness of the importance of FH in health b. give the public tools to gather, understand, evaluate, and use FH to improve their health c. increase awareness of health professionals about the importance of FH d. give health professionals tools to gather, evaluate, and use FH information 2. AMA Website: Family History Tools (http://www.ama-assn.org/ama/pub/category/2380.html) Pedigree exercise. 1. Your patient, John (born in 1984), has glaucoma. You took the family history and found that John has a younger sister (born 1987). John’s mother (born in 1960) has glaucoma. His mother had 4 siblings: an older sister, a younger brother, and fraternal twin sisters (the youngest of the siblings). The brother and one of the youngest sisters also had glaucoma. In addition, the mother’s father had glaucoma. The mother’s mother died in 2001 of a massive MI 2. Construct the pedigree for this case using the pedigree chart handout. Selected Bibliography Bennett RL. The practical guide to the genetic family history. New York: John Wiley, 2008. Wormington CM. How to put genetics into your practice now: Here are 8 ways to incorporate the “new genetic” thinking into your diagnosis and management. Rev Optom 2004:114(11):61-66. Useful Websites AMA Website: Family History Tools, http://www.ama-assn.org/ama/pub/category/2380.html Genetics and Your Practice Website, http://www.marchofdimes.com/gyponline/index.bm2 GeneTests Website, http://www.genetests.org U.S. Surgeon General’s Family History Initiative, http://www.hhs.gov/familyhistory/