FOLDING & UNFOLDING Introduction The process of denaturation
advertisement

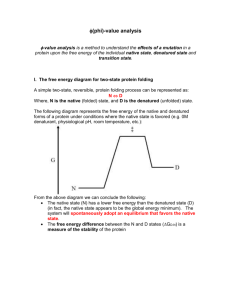
FOLDING & UNFOLDING Introduction The process of denaturation and folding are the two opposite ways through which the specific cellular activity of a protein is created or abolished. The study of the folding and of the denaturation can give useful information about the stability of the protein and the identification of the preferred ways to achieve a specific three-dimensional structure. The prediction of the threedimensional structure of a protein from its amino acid sequence, is perhaps the not resolved most important issue in the modern structural biology. If there was a general solution to the problem of folding it would be possible to write a program for the simulation of the process, avoiding to approach the problem from the experimental point of view. This chapter will deal with these two processes which, although opposite in nature, present a number of common elements. In Figure 1 two states of the same protein, the native and the denatured state, are represented. Figura 1. Equilibrium between the native and the denatured state The two states have a relatively low difference in energy, implying that the two states are separated by an high energy activated state, otherwise we would have a continuous interconversion between the native and the denatured state. On the other hand it is known that the native state, in physiological conditions, is fully stable and the conditions must change dramatically to start the process of denaturation. The transition between the two states can occur through one or more preferential routes, as shown in Figure 2, with formation of one or more intermediates. 97 Figura 2. Possibile folding pathways The information contained in the amino acid sequence of the protein is sufficient to force the protein to reach its three-dimensional structure. This has been demonstrated in the early'70s, when Anfinsen showed that ribonuclease could achieve in vitro its three-dimensional structure starting from the denatured state, once removed the conditions for the denaturation, such as the presence of urea and reducing agents (Fig. 3). The experiment testified for the first time that it was possible in vitro to reversibly fold and unfold a protein. Figura 3. The folding scheme for ribonuclease This process, namely the transition from the denatured to the native form, in vivo can be modulated by a series of molecular factors, called chaperons. Indeed, in vivo there will be proteins that reach the three-dimensional structure regardless of the interaction with the molecular chaperons, while other ones will find more convenient to interact with them. 98 The folding process is fast The process of folding is fast, the protein reaches its native state in less than a few seconds. The process then cannot be completed by sampling all possible conformations followed by the subsequent choice of the structure with the lowest energy, since this procedure would require an extremely long time, as shown in Figure 4. Figura 4. Time required for the folding of a protein with 150 residues. Indeed, in a protein of 150 residues where there are only three possible conformations for each residue and in which the time of interconversion between the conformations is extremely rapid (one picosecond), the total number of conformations are 3150 and the time necessary to sample them would require 1048 years, a time close to infinite when compared to the time experimentally necessary. So the choice of the proper conformation is not through a sampling of all the conformations that can be achieved, but through one or more preferential routes through which the amino acid sequence will define the three dimensional structure which is associated to the function. In Figure 5 ( bottom) a possible outline of the process of folding according to its reaction coordinate is depicted 99 Figura 5. Scheme of the folding process The symbol U defines the protein in the denatured state (unfolded), F in the folded state (folded). These two states are not too much separated in energy but between them there is a state T (the transition), which has a high energy value. This prevents the native and denatured state to stay in a constant equilibrium. M is the protein in the molten globule state, a relatively stable intermediate in the process that goes from U to F, usually characterized by the presence of secondary structure but the lack of a defined tertiary structure. The diagram on the top of figure5 shows the decrease of the distribution of conformations from the denatured protein to the the native form, that, however, still possess a degree of microheterogeneity. The protein folding is a cooperative process. The process of folding is cooperative, a phenomenon due to the simultaneous presence of multiple interactions in a single molecule. Let consider a denatured protein where there are two groups A and B, that can be in contact via a weak interaction. Figura 6. The cooperativity for the interaction of two molecules. The equilibrium constant for the interaction between the two groups in the denatured protein is given by:. 100 KU = KAB[A/B]U where KAB is the association constant of the two groups when they are on separate molecules and [A / B] U is the concentration of the two groups in the denatured protein. The value of [A / B] U ranges from 10-2 to 10-5 M and depends on the relative position of the two groups within the protein. In the presence of multiple interactions the value of concentration can change and thus facilitate or hinder the interaction of groups A and B. If the protein is completely stretched A and B are far apart, when folded A and B are very close and therefore the apparent concentration of A and B is much higher. Basically, the more A and B are close the larger is the apparent concentration of A and B, increasing their interaction possibility. Consider a protein where there are four groups A, B, C and D (Fig.6). The groups can exist in a non interacting, in a partially or fully interacting conformation. The probability of interaction of A and B will be higher in a partially folded than in the denatured conformation. Their association constant KAB remains unchanged (because the groups are always the same), the only thing that changes is the relative distance of the groups. The ratio [A / B] II / [A / B] U, that is the ratio between the apparent concentration of A and B in the partially folded state and in the denatured state represents the cooperativity factor, the factor that facilitates the interaction between A and B. The apparent concentration of A and B is increasing as a result of the interaction between C and D as well as in a symmetric way the interaction of C and D increases as a result of the interaction between A and B. In the case of a protein, once a number of groups are beginning to interact with each other, all others have a greater chance to interact and this means that the process is cooperative. In the event of multiple interactions the final equilibrium constant between the total final state (fully interacting groups) and the initial state ( not interacting state) is given by the following product : KNET = (KAB[A/B]U) (KCD[C/D]I) (KEF[E/F]III) The final state will be stable if KNET will be greater than unity. This is easily understandable considering the presence of multiple groups that can interact with each other, the initial interaction has an equilibrium constant of 10-4 and each additional interaction is 10 times stronger than the previous one. The first interaction will give rise to an equilibrium constant equal to 10-4, the second one equal to 10-7 (10-4 × 10-3 ), the third one 10-9 (10-4 × 10-3 × 10-2 ) and so 101 on. This is because the presence of the first interaction increases the effective concentration of the latter groups and therefore the second interaction will be greater by a factor of 10 which is the cooperativity factor. Initially the product of the various equilibrium constants is always less than one and even the equilibrium constant of the second, third and fourth term is lower than the preceeding one. This process continues until the actual concentration of the interacting groups is such that the equilibrium constant of each interacting group is greater than unity. From this point onwards KNET starts to increase for each subsequent interaction, until the value becomes higher than unity, corresponding to a difference in free energy of zero between the folded and the unfolded form. Figure 7 shows the values of equilibrium constants and the corresponding energy value for a protein in which the equilibrium constant for the first interaction is 10-4 and any subsequent one is 10 times stronger. Figura 7. Energetic scheme for the folding of a molecule where each interaction is 10 times stronger than the previous one. The graph gives rise to a bell curve whose ends represent the completely denatured and the completely interacting groups respectively. The system is stable only in these two points and not in the intermediate states. In Figure 8 the distributions of configurations of a protein during the process of denaturation for a cooperative (left column) or non-cooperative (right 102 column) process are represented. In both cases the protein in the native state is represented by a distribution of conformations relatively narrow, while the denatured protein is represented by a broad distribution of conformations. In the intermediate situation, if the process is cooperative (Fig. 8, left column), there is an equilibrium between two families of conformations represented by the native protein and the denatured protein. If the process is not cooperative, the protein is represented by a broad distribution of conformations, (Fig. 8, right column). Therefore, the cooperativity of the folding process constrains the protein to stay, in partially denaturing conditions, in two well-defined conformations, the native and the denatured state, that are in equilibrium. The K equilibrium constant is the ratio between the percentage of protein in the native state over the percentage of protein in the denatured state and will change varying the external denaturing conditions. Figura 8. Distribution of the protein conformations for a cooperative (left) and non cooperative (right) folding process The presence of an equilibrium between these two forms permits us to measure the difference in free energy between the native and the denatured state and to quantitatively measure for each protein how much the native state is more stable than the denatured one. In fact, for a two states transition K = [N] / [U] and the difference in free energy ΔG between the native state N and the denatured state U is given by:. ΔG = GN – GU = -RTln K To assess this difference in energy the protein must be denatured in a 103 reversibile manner following a parameter diagnostic of the native and of the denatured state of the protein. This will permit to measure the equilibrium constant and thus the energy difference between the two states. How can a protein be denatured? It will be necessary to vary the conditions that maintain the native state. This can be achieved by varying physical (temperature, pressure) or chemical parameters (increased concentration of a denaturant molecule, variation of pH). In fact, the proteins are stable at neutral pH, where it exists a fair balance between the positive and negative charges of the various side chains. A change to acidic or alkaline pH creates a large concentration of negative or positive charges that repel each other leading to the denaturation of the protein itself. One method widely used for the denaturation is to add external molecules that can be divided into two main classes: those that preferentially interact with the protein (preferential binding) and those that prefer to interact with the solvent (preferential hydration), as shown in Figure 9. In general, the molecules that prefer to interact with the solvent are stabilizing factors, while the molecules that prefer to interact with the surfaces of the protein, in particular with the non-polar surfaces, are denaturants. Figure 9 shows on the left molecules that prefer to interact with the protein and on the right molecules that interact preferentially with the water. Figura 9. Distribution of molecules preferentially interacting with the protein (left) or with the water (right). 104 The two chemical denaturants that are most commonly used are urea and guanidine chloride, which are able to increase the solubility of both polar and not polar in a way proportional to the surface accessible to solvent. Figure 10 shows the ΔG of transfer of side chains from water to urea or guanidine chloride. The ΔG is negative and increases in absolute value with the surface of the molecule. This means that the side chains prefer to interact with the denaturant than with water and this is why the protein tends to denature. In fact, the hydrophobic residues at high urea concentrations do not want to be buried in the internal part of a protein, but they want to go to the surface to interact with the denaturant. Figura 10. Free energy of transfer of the aminoacid side chains from water to a denaturing agent. A denaturation curve can be obtained following a parameter, that allows to distinguish the native and the denatured form, as a function of the concentration of a denaturing agent. The curve has a sigmoidal shape, indicative of a cooperative process, as can be seen from Figure 11 where a gel electrophoresis of cytochrome c in urea gradient is reported. 105 Figura 11. Gel electrophoresis of cytochrome c in urea gradient Increasing the urea concentration the protein is more slow as it opens and this allows you to see a separation of the forms. In general, the process of denaturation can be efficiently followed by following a spectroscopic parameter. The emission of fluorescence of a tryptophan may be a good parameter, if the tryptophan in the native state is located within the internal region of the protein. The wavelength of emission of tryptophan depends on its location, being at low wavelength values when it is in a hydrophobic environment and at high values when it is in a hydrophilic environment, as when it is denatured. The selected parameter must have very different characteristics for the protein in the native and in the denatured state. The measures must be taken after the addition of increasing amounts of the denaturantand when we are sure that the equilibrium has been reached, in order to properly assess the percentage of protein molecules in the native and denatured state at that particular concentration of denaturant. In this way, an equilibrium constant can be measured and the difference in free energy ΔG between the native and denatured state can be assessed. After introducing a defined amount of denaturant concentration (1 M, 2 M), the measurement is taken and then repeated as a function of time, until there is no change compared to the previous measurement. This means that the reaction has reached an equilibrium and the corresponding value can be reported in a graph (Fig. 12), where the experimental parameters such as absorbance, fluorescence intensity etc.. are reported as a function of the denaturant concentration. The obtained curves are sygmoidal confirming that the process is cooperative. 106 Figura 12. Denaturation of a protein as a function of the denaturant concentration At the beginning and at the end of the graph in Figure 12, the base line is not perfect though, initially, 100% of protein is present in solution in the native state and at the end, 100% of the molecules have been denatured. The absence of a flat base-line is due to the interaction of the macromolecule with the denaturant, which can cause a variation of the parameter without a disruption of the state of the protein. It is interesting to note that the phenomenon of denaturation occurs in a narrow range of denaturant concentration or temperature, as shown in Figure 12 and 12b. Figura 12b. Protein denaturation as a function of temperature. In this interval the parameter changes rapidly, because its value depends on the number of molecules that pass from the native to the denatured state. In this range the protein is not in an undefined state, but the solution contains only two conformations: the native and the denatured state. The experimental measured parameter has a value that is given by the linear combination of the contribution of molecules in the native state plus the contribution of the molecules in the denatured state. This allows you to measure for each denaturant concentration an equilibrium constant given by the ratio between the percentage of molecules in the native and the denatured state. It is in fact 107 possibile to write a system of two equations and two unknowns, represented by the fraction of molecules in native state fF and the fraction in the denatured state fU (Fig. 13). The sum of the two fractions must be constant and equal to 1 and the experimental parameter y (ie the experimental parameter measured as a function of the concentration of denaturant) is due to the contribution of the value of the parameter for the native form yF multiplied by the native fraction fF and the value for the denatured form yU multiplied by the denatured fraction fU Figura 13. The two equations necessary to work out the free energy difference between the native and the denatured state. The resolution of the system provides the equilibrium constant K for each value of the concentration of denaturant, so a constant K1, or a constant K2, K3 for three different urea concentrations. The differences in free energy ΔG1, ΔG2, ΔG3 corresponding to these equilibrium constant values can be reported on a graph having as abscissa the concentration of denaturant and as ordinate the values of free energy changes. Which is the ΔG value indicative of the energy difference between the native and the denatured protein? The most appropriate value is the value that occurs in the absence of denaturant but in these conditions the protein is in the native state and it is therefore not possible to measure the equilibrium constant. This value can be obtained by extrapolation. The graph of the ΔG values as a function of urea concentration is a straight line whose intercept with the y axis for the value of x = 0 is a good approximation of the value of the free energy difference between the native and denatured state of the protein at physiological conditions. In Figure 14 the value is 6 kcal / mol, corresponding to the contribution of 2 or 3 hydrogen bonds. The two states are so relatively close in energy that should be in equilibrium, but this does not happen because they are separated by an intermediate state, the transition state, that is characterized by a high energy value, so it is necessary to strongly vary the environmental conditions to denature the protein. The energy difference ΔG between the native and the denatured state for any globular protein, is of the same order of magnitude that is typically less than 10 Kcal / mol 108 Figura 14. Free energy differente between the native and the denatured state as a function of urea concentration. The dependence of the thermodynamic parameters for the native and the denatured state as a function of temperature shows that the enthalpy and entropy changes are large and in both cases of the same order of magnitude, of hundreds of kcal / mol (Fig. 15) . The corresponding free energies are of a few tens of kcal / mol and the same goes for the free energy difference between the native and denatured. The graph in Figure 15 shows an example of a protein in which up to a temperature of 80 ° C the native state is more stable the denatured one, and after this temperature, the opposite happens. Figura 15.Dependence on temperature of the entropic and entalpic parameter The graph in Figure 16, that shows the difference in free energy ΔG between the native and denatured state as a function of temperature for a series of proteins, 109 indicates that each protein has an optimal stability temperature. The graph has a bell shape, indicating that not all proteins like a very low temperature. Figura 16. Free energy difference of the native and denatured state as a function of temperature. Qualitative comparison of the stability of a native and mutated protein In the preceding paragraph it has been shown how to quantitatively assess the difference in stability between the native and denatured state of a protein through a reversible denaturation that allows us to assess the relative thermodynamic parameters. The qualitative measure of the relative stability of a native protein in comparison to a mutant does not require a reversible denaturation and informations can be obtained more directly. In this case it is possible to obtain a scale of relative stability by making an irreversible denaturation, bringing for example the protein at 100 ° C, maintaining this temperature for a definite time and then bringing it to room temperature. When the temperature is increased to 100 ° C, an irreversible denaturation occurs. It is possibile to measure the percentage of denaturation comparing the activity before and after the incubation at 100 ° C. This procedure, performed in a comparative way on both the native and the mutated protein, allows us to understand whether the mutant is more or less stable than the native protein. In fact in a graph representing the ln of the activity as function of the incubation time at high temperature it is possible to assess the speed of the activity loss. In the graph in Figure 17, the loss of the activity of the mutant represented by white ball is slower than the native (black dot). This means that the mutation is a stabilizing mutation, while the mutation represented by the diamond is destabilizing because the protein is leaking faster the activity. It is enough to 110 irreversibly denaturate the proteins to define a level of stability and determine the stabilizing mutations.. Figura 17. Graph of the ln of the activity as a function of the incubation time at high temperature for the native and mutated samples If this type of experiment is performed with dimeric or multimeric proteins it may be useful to do the measurements as a function of enzyme concentration. In this way you can understand if the path followed during the denaturation is a transition from the dimeric form directly to the denatured state (DU) or from the dimer to the monomer and finally to the denatured state (DMU). If the loss of activity is concentration dependent, it means that the path followed is DMU because lowering the concentration the monomer-dimer equilibrium is shifted toward the monomer and the protein tends be more easily denaturated. Instead, if it is concentration independent, the route followed is DU as the rate of denaturation doesn’t depend on the -dimer-monomer equilibrium (Fig. 18).. Figura 18. Rate of the loss of activity as a function of protein concentration for a dimeric enzyme following the DMU path. Stabilizing mutations 111 It is possible to make mutations that render the enzymes more stable. Nearly all consist in the introduction of a new interactions within the folded protein (Fig. 19). Figura 19. Possibili strategie per stabilizzare le proteine. A particularly effective strategy is to introduce new disulfide bridges. The succesfull case, however, are relatively rare because the disulfide bridges have very specific geometric constraints and it is not so easy to introduce them by mutagenesis. It is necessary to know the three-dimensional structure of the protein in order to engineer the cysteines in the right position so that the disulfide bridge will occur. Another strategy consists in introducing new residues that form electrostatic interactions or hydrogen bonds. For example, the introduction of a negative charge at the N-terminal of an α helix leads to a stabilization due to the presence of the dipole moment of the helix. The introduction of binding sites for metal ions is another stabilizing strategy. The resolution of threedimensional structure of thousands of proteins has made possible the description of many metal sites, the identification of their favorite ligands, the preferred distance and geometry of this bond and allows the introduction of specific aminoacids such as histidine, aspartate, cysteine, which are able to efficiently bind atoms of copper, zinc or other metals and stabilize the protein. The mutations above described introduce new interactions and thus stabilize the protein for enthalpic reasons. The introduction of proline residues and the replacement of glycine residues instead stabilize the protein for entropic reasons. In fact in the native state both glycines and prolines have specific values for the Φ and Ψ angles defined by the constraints of the whole protein. In the denatured state, however, the glycine can sample a conformational space much higher than proline. This is due to the fact that the proline has a low conformational freedom, whereas glycine is the amino acid that has the greatest conformational freedom. Thus, a protein with a high number of glycines tends towards the denatured state for entropic reasons, because in the denatured state glycine can play the 112 conformational freedom that cannot in the folded state because of the constraints imposed by the rest of the protein. In particular, substitution of glycines with prolines, the amino acid characterized by the lowest degree of conformational freedom, leads to a strong stabilization for entropic reasons. The effect is produced either by introducing prolines or eliminating glycines. 113