Most suitable methods for strain identification
advertisement

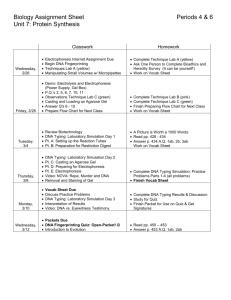
SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 For SCAMDM comments and approval for publication Version including agreed PG comments Guidance Document Identification of probiotics at strain level Knut J. Heller1, W. Bockelmann1, E. Brockmann2 Max Rubner-Institut (Federeal Research Institute for Nutrition and Food), Department of Microbiology and Biotechnology, Hermann-Weigmann-Str. 1, D-24103 Kiel, Germany; 2 Chr. Hansen, Bøge Allé 10-12, DK-2970 Hørsholm, Denmark *Corresponding author: Email <knut.heller@mri.bund.de> 1 Probiotic properties are those of strains, not of species or even higher taxa (Heller, 2003). This has been stressed by WHO/FAO in 2002 (N.N., 2006) by the following statement: “Strain typing has to be performed with a reproducible genetic method or using a unique phenotypic trait. Pulsed Field Gel Electrophoresis (PFGE) is the gold standard. Randomly Amplified Polymorphic DNA (RAPD) can also be used, but is less reproducible. Determination of the presence of extrachromosomal genetic elements, such as plasmids can contribute to strain typing and characterization.” That probiotic properties are those of strains and not of higher taxa is reflected by the fact that species names of micro-organisms with established probiotic properties are extended by additional identifiers: e.g. a combination of letters and numbers or the name of the person who isolated it. Examples are: Lactobacillus rhamnosus GG, Bifidobacterium animalis subsp. lactis BB-12, Lactobacillus casei Shirota, etc. In addition to strain-specific methods needed for identification of probiotic strains, correct assignment to species or sub-species remains indispensable. The latter is important for evaluating the genetic background probiotic micro-organisms are imbedded in: whether they belong to groups with established positive impacts on human health or whether they belong to groups which include spoilage or even pathogenic micro-organisms (Pot et al., 1997). In this communication we focus on strain identification. However, it has to be clear that this identification is just based on a typing method, which only allows to demonstrate identity or non-identity of a strain in question with that of a given probiotic strain in the context/frame of the typing scheme of the applied method. Typing methods have been compared in several reviews (Vandamme et al., 1996; Salvelkoul et al., 1999; Domigk et al., 2003) and they have been discussed with respect to their taxonomic resolution powers. A consensus of the latter is represented in Fig. 1. In a rather recent review, Li et al. (2009) have focussed on genomic typing methods and have extended the overview by including methods based solely on DNA extracted from the environment. The methods with the broadest range of application are “DNA hybridization probes” and “DNA sequencing”. They basically allow taxonomic discrimination from the highest down to the lowest level, the strain level. However, while many “DNA hybridization probes” have been developed and published for higher taxonomic levels (targeting rDNA), only few are available for strain levels (targeting genes specific for the strains). The reasons for this are rather simple: DNA probes for strain level identification have very limited applications but require intensive and laborious testing for their development. DNA sequencing, on the other hand, has the potential to become the standard method for all problems of taxonomic resolution, even when considering resolution below species level. At present, sequencing of ribosomal RNA genes (16S rDNA) represents the standard for species identification, since standard amplification primers binding to conserved regions and standard sequencing primers 1 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 can be applied. For strain identification, however, 16S rDNA sequencing cannot be successfully applied, due to limited variability of this gene. One has to acknowledge that very powerful sequencing techniques have been developed during recent years, which allow sequencing of large numbers of nucleotides at rather low cost. This has led to the development of the “Multi Locus Sequencing Typing / Multi Locus Sequence Analysis (MLST / MLSA)” technique (Maiden et al., 1998), in which several gene loci are sequenced and the sequence information generated is catenated and used for differentiation and typing purposes. During recent years this technique has gained momentum and several MLST databases have been developed and are accessible online. The most recently developed “next generation” sequencing techniques, the “454 sequencing” (Margulies et al., 2005) and the “Solexa/Illumina” and “SOLiD” sequencing, bear the potential of sequencing and analysing entire genomes of microorganisms within one or a few days. Thus, whole genome sequencing may eventually develop into the standard method of strain typing at least for industrially important strains. However, before whole genome sequencing can become a standard typing method, a clear concept has to be developed for defining the extent of allowable sequence deviation within one and the same strain. For very obvious reasons, “phenotype determination by classical methods” is not an appropriate method for taxonomic resolution at strain level: it simply requires too much time and experimental efforts. Many other methods listed in Fig. 1, e.g. rRNA sequencing, cell wall structure, % G+C, are not suitable for discriminating at strain level, since discrimination is limited to higher taxa. These methods will not be discussed any further. Of the methods suitable for strain identification, those based on serology target the surfaces of micro-organisms. Serology has been very successfully applied for differentiation of pathogenic bacteria. However, so far it has not been shown whether serology is capable of identifying a probiotic strain among its non-probiotic relatives within the same species. SDS-PAGE (sodium dodecylsulphate polyacrylamide gel electrophoresis) of total cellular protein separated basically according to size (molecular mass) has been successfully shown to be applicable in lactic acid bacteria for differentiation at strain level (Pot et al., 1997). However, due to the necessity of growing and harvesting the cells under strictly defined conditions for generation of absolutely reproducible protein patterns, this method is powerful when used as “in-house” method. However, for identification of strains in different laboratories the method may not be robust enough. Similar considerations regard DNAamplification techniques, where amplification conditions are applied, which are based on primers and primer binding conditions allowing to target sequences not exactly matching the primer sequences. This is especially true for RAPD (random amplification of polymorphic DNA), where primers with arbitrary sequences are applied, and to a somewhat lesser extent for rep-PCR (Repetitive Element PCR) and other similar methods like ERIC-PCR (Enterobacterial Repetitive Intergenic Consensus PCR) (de Bruijn, 1992), Box-PCR (BOXA1R-based repetitive extragenic palindromic PCR) (Louws et al., 1994) etc. In these cases small changes in the amplification procedures result in significant changes in the electrophoretic patterns generated. As a consequence, inter-laboratory identification at strain level becomes very difficult if not impossible. Somewhat reduced robustness is also the major argument against AFLP (amplified fragment length polymorphism) (Vos et al., 1995), which otherwise is a method of very high discriminating power and which allows high throughput of samples to be tested. In this method, genomic DNA is hydrolysed by two restriction enzymes of which at least one is a frequent cutter enzyme. To the ends generated, two different adapters are ligated, which differentially recognize the two different ends produced by the two enzymes. For subsequent PCR, primers basically corresponding to the adapter sequences but with extended selectivity are applied. 2 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 In contrast to AFLP, ribotyping (Snipes et al., 1989) and PFGE (Snell and Wilkins, 1986) do not involve any step of amplification by PCR. For ribotyping, chromosomal DNA is hydrolysed by restriction enzymes and separated by agarose gel electrophoresis. In the following Southern blot, a labelled (radioactive, fluorescence, digoxigenin etc.) probe specific for rRNA genes identifies those DNA fragments, which carry regions of those rRNA genes. Due to the larges heterogeneity of DNA regions flanking the rRNA genes, the banding patterns of hybridizing fragments show very high intra-species variation. In PFGE, chromosomal DNA is hydrolyzed by means of a rare cutting restriction enzyme. The very large DNA fragments obtained are separated by an electrophoresis technique, in which the electric field frequently changes between two different directions, which leads to clear separation of DNA fragments several hundred thousand basepairs in length. This leads to typical banding patterns , which are used for fingerprinting. Thus, both ribotyping and PFGE are very robust techniques, since exactly defined, reproducible electrophoresis patterns are generated by complete hydrolysis of DNA with restriction endonucleases: ambiguities resulting from varying amplification conditions are thus excluded. However, ambiguities with this technique may arise from poor hydrolysis of bacterial cell walls for liberation of DNA within the plug moulds, incomplete restriction hydrolysis, and variations in the electrophoresis conditions. The latter, however, can be compensated for by always using defined marker-DNA as control in the electrophoretic separation. Actually, for epidemiologic studies, PFGE has been demonstrated to be applicable as a standardized effective method for identification of strains in foodborne disease outbreaks by concerted actions of laboratories joined in the so-called Pulse-Net (Boxrud et al., 2010). Some of the typing techniques have been compared with each other with respect to their discriminating powers. In a survey involving 35 isolates of Campylobacter jejuni, Männinen et al. (2001) discriminated 8 different strains by ribotyping, 10 by PFGE and 10 by AFLP. Tynkkynen et al. (1999) analysed 19 Lactobacillus rhamnosus isolates and were able to discriminate 7 different strains by RAPD, 10 by ribotyping, and 12 by PFGE. Finally, Mättö et al. (2004) analysed 18 Bifidobacterium longum and 10 Bifidobacterium adolescentis isolates. They were able to discriminate for B. longum 7 different strains by RAPD, 13 by ribotyping, and 14 by PFGE (only 14 of the 18 isolates were analysed by PFGE). For B. adolescentis, RAPD yielded 6, and ribotyping as well as PFGE 9 different strains each. Thus, PFGE is a robust method with the best discriminative power at strain level of all methods – except AFLP (Vogel et al., 2004) - described. It is more labour-intensive than e.g. AFLP, but less labour-intensive than ribotyping (a rapid PFGE method for analysis of bifidobacteria has been described some years ago) (Briczinski and Roberts, 2006)). For these reasons, PFGE is called the “gold-standard” of strain identification. This has been acknowledged by FAO/WHO as already indicated in the first paragraph of this document (N.N., 2006). In the Annex I to this Document, the most suitable methods for strain identification are listed together with short descriptions of the methods. The literature cited in Annex I is included in the list of references at the end of the body of this text. The availability of techniques for fingerprinting below species level allows for strain differentiation. However, when applying such techniques, one always has to bear in mind that fingerprinting methods are just typing methods: they analyse one characteristic trait, which then is used for attributing the organisms to groups of identical or very similar organisms (strains) or non-identical ones. Except for methods based on DNA sequencing, typing methods are not suited for phylogenetic considerations. The results of the typing methods for different strains within one species have to be interpreted with care. It has to be clear from the beginning of an experiment, whether any observed deviation from a given pattern is supposed to result in the denomination of a new strain or not. Since strains do 3 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 not form a taxonomic unit but form a group of members, which have been assigned to the same strain on the basis of an arbitrary definition, the definition may be such that it either does not allow for any deviations in a banding pattern or that it allows for small deviations of up to 10% in a banding pattern of one and the same strain. The latter definition is often applied, when tracing back microorganisms in outbreaks to the original source of infection (Chiou et al., 2001). When just one method is applied for assigning microorganisms to one group, one has to be aware, that the group identified by this method may in fact be somewhat heterogeneous. Recently, it has been described that Lactobacillus fermentum strains with identical PFGE patterns differed in up to four characteristic traits (ARDRA pattern generated with a particular restriction enzyme, growth at a particular temperature, ability to metabolize two different sugars) (Njeru et al., 2010). This certainly raises the fundamental question of whether typing methods are really useful in attributing microorganisms to groups of microorganisms sharing one or few important functional traits, which are not tested by the typing method. The Joint FAO/WHO Working Group Report on Drafting Guidelines for the Evaluation of Probiotics in Food (N.N., 2006) states that “Strain identity is important to link a strain to a specific health effect as well as to enable accurate surveillance and epidemiological studies”, thereby indicating that strain identification serves different purposes. High resolving typing methods like PFGE will be adequate for the assessment of recovery in studies investigating probiotic functionality as well as safety, where the strain identity of the probiotic applied is typically not only secured by typing methods but also through a documented origin traceable to a reference material. Due to the described possible heterogeneity of strains belonging to one typing group, typing methods can however not be fully sufficient to generally make the link to probiotic functionality. The same argument will also limit their applicability for accurate surveillance as well as epidemiological studies where there is no documented link to the probiotic reference material. The conclusion for probiotic strains can only be that typing methods like PFGE are important as long as specific tests for those genes – or even for the activity of those genes - involved in making a strain probiotic are not available, due to lack of information of the genes involved. As soon as such information is available, typing methods together with methods testing the activities of relevant genes will show that certain functional traits are present within the correct strain, i.e. in a microorganism with the correct genetic background. References Bennasar, A., Mulet, M., Lalucat, J., García-Valdés, E. 2010. PseudoMLSA: a database for multigenic sequence analysis of Pseudomonas species. BMC Microbiol. 10:118. Briczinski, E.P., Roberts, R.F. 2006. Technical Note: A rapid pulsed-field gel electrophoresis method for analysis of bifidobacteria. J. Dairy Sci. 89:2424-2427 Boxrud, D., Monson, T., Stiles, T., et al. 2010. The role, challenges, and support of Pulse-Net laboratories in detecting foodborne disease outbreaks. Publ. Health Rep. 125:57-62 Busconi, M., Reggi, S., Fogher, C. 2008. Evaluation of biodiversity of lactic acid bacteria microbiota in the calf intestinal tracts. Antonie Van Leeuwenhoek 94(2):145-155. de Bruijn, F.J. 1998. Use of repetitive (Repetitive Extragenic Palindromic and Enterobacterial Repetitive Intergeneric Consensus) sequences and the polymerase chain reaction to fingerprint the genomes of Rhizobium meliloti isolates and other soil bacteria. Appl. Environ. Microbiol. 58:2180-2187 4 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 Chiou, C.S., Hsu, W.B., Wei, H.L., Chen, J.H. 2001. Molecular epidemiology of a Shigella flexneri outbreak in a mountainous township in Taiwan, Republic of China. J. Clin. Microbiol. 39:1048-1056 De Vuyst, L., Vancanneyt, M. 2007. Biodiversity and identification of sourdough lactic acid bacteria. Food Microbiol. 24(2):120-127. Domigk, K.J., Mayer, H.K., Kneifel, W. 2003. Methods used for the isolation, enumeration, characterisation and identification of Enterococcus spp. Int. J. Food Microbiol. 88:165188 Engel, G., Roesch, N., Heller, K.J. 2003. Typing by pulsed-field gel electrophoresis (PFGE) of bifidobacteria from fermented dairy products. Kieler Milchw. Forsch. 55:225–232 (in German) Hanage, W.P., Fraser, C., Spratt, B.G. 2006. Sequences, sequence clusters and bacterial species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361(1475):1917-1927. Hänninen, M.-L., Perko-Mäkelä, P., Rautelin, H., Duim, B., Wagenaar, J. A. (2001). Genomic Relatedness within Five Common Finnish Campylobacter jejuni Pulsed-Field Gel Electrophoresis Genotypes Studied by Amplified Fragment Length Polymorphism Analysis, Ribotyping, and Serotyping. Appl. Environ. Microbiol. 67:1581-1586. Heller, K.J. 2003. Inclusion of Probiotics in Beverages: Can it Lead to Improved Health? pp. 247-257. In T. Wilson and N. J. Temple (Eds.), Beverages in Nutrition and Health. Humana Press Inc., Totowa, New Jersey. Hudson, M.E. 2008. Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol. Ecol. Resour. 8(1):3-17. Klein, G., Pack, A., Bonaparte, C., et al. 1998. Taxonomy and physiology of probiotic lactic acid bacteria. Int. J. Food Microbiol. 41:103-125 Li, W., Raoult, D. Fournier, P.-E. 2009. Bacterial strain typing in the genomic era. FEMS Microbiol. Rev. 33:892–916 Lortal, S., Rouault, A., Guezenec, S., Gautier, M. 1997. Lactobacillus helveticus: strain typing and genome size estimation by pulsed field gel electrophoresis. Curr. Microbiol. 34:180-185 Louws, F.J., Fulbright, D.W., Taylor-Stephens, C., de Bruijn, F.J. 1994. Specific Genomic Fingerprints of Phytopathogenic Xanthomonas and Pseudomonas Pathovars and Strains Generated with Repetitive Sequences and PCR. Appl. Environ. Microbiol. 60:22862295 Maiden, M.C.J., Bygraves, J.A., Feil, E. et al. 1998. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms Proc. Natl. Acad. Sci. U.S.A. 95: 3140–3145 Margulies, M. Egholm, M. Altman, W.E. et al., 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376-380 Mättö, J., Malinen, E., Suihko, M.-L., Alander, M., Palva, A., Saarela, M. 2004. Genetic heterogeneity and functional properties of intestinal bifidobacteria. J. Appl. Microbiol. 97:459-462 Möller, C. 2002. Application of new selection strategies for yoghurt starter cultures suitable for yielding yoghurt with mild taste. Ph.D. thesis, University at Kiel, Germany (in German) 5 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 N.N. 2006. Joint FAO/WHO Working Group Report on Drafting Guidelines for the Evaluation of Probiotics in Food. FAO Food and Nutrition Paper 85 Njeru, P.N., Roesch, N., Ghadimi, D., et al., 2010. Identification and characterization of lactobacilli isolated from 1 Kimere, a spontaneously 2 fermented pearl millet dough from Mbeere, Kenya (East Africa). Beneficial Microbes (in press) Pennacchia, C., Vaughan, E.E., Villani, F. 2006. Potential probiotic Lactobacillus strains from fermented sausages: Further investigations on their probiotic properties. Meat Sci. 73: 90-101 Picozzi, C, Bonacina G, Vigentini I, et al. 2010. Genetic diversity in Italian Lactobacillus sanfranciscensis strains assessed by multilocus sequence typing and pulsed-field gel electrophoresis analyses. Microbiology-SGM 156: 2035-2045 Pot, B., Coenye, T, Kersters, K. 1997. The taxonomy of microorganisms used as probiotics with special focus on enterococci, lactococci, and lactobacilli. Microecology and Therapy, 26: 11-25 Rong, X., Liu, N., Ruan, J., Huang, Y. 2010. Multilocus sequence analysis of Streptomyces griseus isolates delineating intraspecific diversity in terms of both taxonomy and biosynthetic potential. Antonie van Leeuwenhoek 98:237–248 Roy, D., Ward, P., Champagne, G. 1996. Differentiation of bifidobacteria by use of pulsedfield gel electrophoresis and polymerase chain reaction. INTERNATIONAL JOURNAL OF FOOD MICROBIOLOGY 29: 1:11-29 Savelkoul, P.H., Aarts, H.J., de Haas, J., Dijkshoorn, L., Duim, B., Otsen, M., Rademake,r J.L., Schouls, L., Lenstra, J.A. 1999. Amplified-fragment length polymorphism analysis: the state of an art. J. Clin. Microbiol. 37:3083-91 Scheirlinck, I., Van der Meulen, R., De Vuyst, L., Vandamme, P., Huys, G. 2009. Molecular source tracking of predominant lactic acid bacteria in traditional Belgian sourdoughs and their production environments. J. Appl. Microbiol. 106(4):1081-92. Snell RG, Wilkins RJ. 1986. Separation of chromosomal DNA molecules from C.albicans by pulsed field gel electrophoresis. Nucleic Acids Res. 14:4401-4406 Snipes, K.P., Hirsh, D.C., Kasten, R.W. et al.1989. Use of an rRNA probe and restriction endonuclease analysis to fingerprint Pasteurella multocida isolated from turkeys and wildlife. J. Clin. Microbiol.27:1847-1853 Tynkkynen, S., Satokari, R., Saarela, M., Mattila-Sandholm, T., Saxelin, M. 1999. Comparison of ribotyping, randomly amplified polymorphic DNA analysis, and pulsedfield gel electrophoresis in typing of Lactobacillus rhamnosus and L. casei strains. Appl. Environ. Microbiol. 65:3908-14 Vancanneyt, M., Huys, G., Lefebvre, K., Vankerckhoven, V., Goossens, H., Swings, J. 2006. Intraspecific genotypic characterization of Lactobacillus rhamnosus strains intended for probiotic use and isolates of human origin. Appl. Environ. Microbiol. 72:5376-83 Vandamme, P., Pot, B., Gillis, M., De Vos, P., Kersters, K., Swings, J. 1996. Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol Rev. 60:407–438 Ventura, M., Zink, R. 2002. Rapid identification, differentiation, and proposed new taxonomic classification of Bifidobacterium lactis. Appl. Environ. Microbiol. 68: 64296434 Vogel, B., Fussing, V., Ojeniyi, B., Gram, L., Ahrens, P. 2004. High-resolution genotyping of Listeria monocytogenes by fluorescent amplified fragment length polymorphism 6 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 analysis compared to pulsed-field gel electrophoresis, random amplified polymorphic DNA analysis, ribotyping, and PCR-restriction fragment length polymorphism analysis. J. Food Prot. 67:1656-1665 Vos, P., Hogers, R., Bleeker, M., et al. 1995 AFLP – a new technique for DNA-fingerprintig. Nucl. Acids Res. 23:4407-4414 7 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 FAMILY GENUS SPECIES STRAIN DNA probes, DNA sequencing rDNA sequencing phenotype (classical, API, Biolog etc.) Cell wall structure Cellular fatty acid fingerprinting (FAME) % G+C DNA-DNA hybridization Phage and bacteriocin typing DNA amplification (ARDRA) SDS-PAGE patterns Serological methods DNA amplification (AFLP, RAPD, rep-PCR etc.) Ribotyping Restriction fragment length polymorphism (RFLP, PFGE) Fig. 1: Taxonomic resolution of different typing techniques. The figure was created based on information presented in Vandamme et al. (1996), Salvelkoul et al. (1999), Domigk et al. (2003). 8 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 Annex I Most suitable methods for strain identification DNA sequencing-based methods Due to extreme advances in development of DNA sequencing methodology, DNA sequencing-based methods offer great advantages for typing at all phylogenetic levels, including strain level. An update on typing methods in the genomic era has been published by Li et al. (2009). DNA sequencing, in contrast to other typing methods, bears the advantage of simultaneously comprising typing and phylogenetic classification. - MLSA/MLST (Multi Locus Sequence Analysis/Multi Locus Sequence Typing) MLSA/MLST is a technique, which has been developed during recent years (Maiden et al., 1998). In this technique, several gene loci are sequenced and the sequence information generated is catenated and used for differentiation and typing purposes. The gene loci chosen usually are house-keeping genes (Hanage et al., 2006), like atpD, gyrB, recA, rpoB, rpoD, etc., which may be combined with the 16S and 23S rRNA genes (Bennasar et al., 2010), which are rather conserved. Inclusion of the rRNA genes allows for inclusion of all phylogenetic data produced so far for species identification. Application of MLSA has been described for lactic acid bacteria populations in sourdough (De Vuyst and Vancanneyt, 2007). While the concept for applying MLSA/MLST for species identification is rather straightforward and clear, this does not hold true for strain identification. It is by no means clear, how much sequence divergence would be allowed to still cluster isolates within the same strain. - Whole genome sequencing Especially the most recently developed “next generation” sequencing techniques, the “454 sequencing” (Margulies et al., 2005) and the “Solexa/Illumina” and “SOLiD” sequencing (Hudson, 2008), bear the potential of generating raw sequence data for entire genomes of microorganisms within less than one day. The powers of these techniques rely on their abilities to generate millions to billions of small sequence reads at one time and thus to produce as much as several gigabases of DNA sequences per run. These techniques have reduced the cost per sequenced base to less than 0.0001 € per base of final sequence and thus offer the possibility to sequence quite a large number of isolates of one species in short time with rather little financial effort. The microbial genome sequences generated can be compared and – as already described for MLSA/MLST sequencing – can be applied for typing and phylogenetic purposes. However, although it should be rather easy to identify organisms belonging to one species (on the basis of the 16S and 23S rRNA gene sequences), a concept for assigning members of one species to a certain strain is still lacking. AFLP (Amplified Fragment Length Polymorphism) Of the PCR-based methods, AFLP (Vos et al., 1995) is the most promising for strain identification. In this method, three steps are involved: i) genomic DNA is hydrolysed by two restriction enzymes of which at least one is a frequent cutting enzyme. To the restriction fragment ends generated, two different adapters are ligated, which differentially recognize the two different ends produced by the two enzymes. ii) For PCR, two different primers are applied, the sequences of which correspond to the respective adapter sequences. However, the primer sequences are extended by at least one additional nucleotide at their 3’-ends. Thus, selectivity of the primers is increased and amplification 9 120531 SCAMDM Meeting 7 June 2012 Tel Aviv IL Agenda item 4 D03 is restricted to maximally 1/16 of all potential possibilities. ii) The amplification products are finally separated on denaturing polyacrylamide gels, the same type used for DNA sequencing. Typically, 50-100 restriction fragments are amplified and detected. Due to the accuracy of the sequencing gels, the pattern generated are very exact and reproducible. The robustness of the method is usually very good, since primer binding takes place at exactly matching complementary sequences. The application of AFLP in the analysis of complex populations of lactic acid bacteria has been described (Busconi et al., 2008; Scheirlinck et al., 2009). Ribotyping For ribotyping, chromosomal DNA, hydrolysed by restriction enzymes and separated by agarose gel electrophoresis, is subjected to a Southern blot and probed with labelled (radioactive, fluorescence, digoxigenin etc.) probes specific for rRNA genes (Snipes et al., 1989). Thereby, all DNA restriction fragments are identified, which carry DNA regions complementary to the probes applied. Due to the large heterogeneity of DNA regions flanking the rRNA genes, the banding patterns of hybridizing fragments show very high intra-species variation. The discriminatory power below species level almost matches that of PFGE, when lactobacilli or bifidobacteria were analysed (Tynkkynen et al., 1999; Mättö et al., 2004). However, ribotyping – although robust - is a very labour and cost intensive technique. PFGE (Pulsed-Field Gel Electrophoresis) As with ribotyping, PFGE does not involve any DNA amplification step, thought to be the critical step when robustness of a technique is considered. For PFGE, chromosomal DNA is hydrolyzed by means of a rare cutting restriction enzyme. The very large DNA fragments obtained are separated by an electrophoresis technique, in which the electric field frequently changes between two different directions. This leads to clear separation of DNA fragments up to several hundred thousand basepairs in length. The banding patterns obtained are used for fingerprinting. Restriction endonucleases recommended have been described in the literature and successfully applied in our laboratory: XbaI and SpeI for bifidobacteria (Roy et al., 1996; Ventura and Zink, 2002; Engel et al., 2003) and AscI (Möller 2002; Vancanneyt et al., 2006; Njeru et al., 2010) and NotI (Pennacchia et al., 2006; Vancanneyt et al., 2006) for lactobacilli. Other enzymes, like e.g. SmaI (Lortal et al., 1997; Klein et al., 1998) and ApaI (Picozzi et al., 2010) have been shown to yield more satisfying results for Lactobacillus species with a low GC-content. In contrast to other techniques, there exists a clear concept for strain identification, since PFGE is the technique widely applied for tracking back foodborne outbreaks due to pathogenic strains (Boxrud et al., 2010). 10 120531