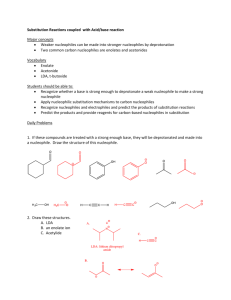
Substitution Reactions
advertisement

Substitution Reactions The functional group of alkyl halides is a carbon-halogen bond, the common halogens being fluorine, chlorine, bromine and iodine. With the exception of iodine, these halogens have electronegativities significantly greater than carbon. Consequently, this functional group is polarized so that the carbon is electrophilic and the halogen is nucleophilic, as shown in the drawing on the right. Two characteristics other than electronegativity also have an important influence on the chemical behavior of these compounds. The first of these is covalent bond strength. The strongest of the carbonhalogen covalent bonds is that to fluorine. Remarkably, this is the strongest common single bond to carbon, being roughly 30 kcal/mole stronger than a carbon-carbon bond and about 15 kcal/mole stronger than a carbon-hydrogen bond. Because of this, alkyl fluorides and fluorocarbons in general are chemically and thermodynamically quite stable, and do not share any of the reactivity patterns shown by the other alkyl halides. The carbon-chlorine covalent bond is slightly weaker than a carbon-carbon bond, and the bonds to the other halogens are weaker still, the bond to iodine being about 33% weaker. The second factor to be considered is the relative stability of the corresponding halide anions, which is likely the form in which these electronegative atoms will be replaced. This stability may be estimated from the relative acidities of the H-X acids, assuming that the strongest acid releases the most stable conjugate base (halide anion). With the exception of HF (pKa = 3.2), all the hydrohalic acids are very strong, small differences being in the direction HCl < HBr < HI. Substitution and Elimination The characteristics noted above lead us to anticipate certain types of reactions that are likely to occur with alkyl halides. In describing these, it is useful to designate the halogen-bearing carbon as alpha and the carbon atom(s) adjacent to it as beta, as noted in the first four equations shown below. Replacement or substitution of the halogen on the α-carbon (colored maroon) by a nucleophilic reagent is a commonly observed reaction. Also, since the electrophilic character introduced by the halogen extends to the β-carbons, and since nucleophiles are also bases, the possibility of base induced H-X elimination must also be considered. Finally, there are some combinations of alkyl halides and nucleophiles that fail to show any reaction over a 24 hour period. For consistency, alkyl bromides have been used in these examples. Similar reactions occur when alkyl chlorides or iodides are used, but the speed of the reactions and the exact distribution of products will change. 20 In order to understand why some combinations of alkyl halides and nucleophiles give a substitution reaction, whereas other combinations give elimination, and still others give no observable reaction, we must investigate systematically the way in which changes in reaction variables perturb the course of the reaction. The following general equation summarizes the factors that will be important in such an investigation. One conclusion, relating the structure of the R-group to possible products, should be immediately obvious. If R- has no beta-hydrogens an elimination reaction is not possible, unless a structural rearrangement occurs first. The first four halides shown on the left below do not give elimination reactions on treatment with base, because they have no βhydrogens. The two halides on the right do not normally undergo such reactions because the potential elimination products have highly strained double or triple bonds. It is also worth noting that sp2 hybridized C–X compounds, such as the three on the right, do not normally undergo nucleophilic substitution reactions, unless other functional groups perturb the double bond(s). .When several reaction variables may be changed, it is important to isolate the effects of each during the course of study. In other words: only one variable should be changed at a time, the others being held as constant as possible. For example, we can examine the effect of changing the halogen substituent from Cl to Br to I, using ethyl as a common R–group, cyanide anion as a common nucleophile, and ethanol as a common solvent. We would find a common substitution product, C2H5–CN, in all cases, but the speed or rate of the reaction would increase in the order: Cl < Br < I. This reactivity order reflects both the strength of the C–X bond, and the stability of X(–) as a leaving group, and leads to the general conclusion that alkyl iodides are the most reactive members of this functional class. 1. Nucleophilicity Recall the definitions of electrophile and nucleophile: 21 Electrophile: An electron deficient atom, ion or molecule that has an affinity for an electron pair, and will bond to a base or nucleophile. Nucleophile: An atom, ion or molecule that has an electron pair that may be donated in forming a covalent bond to an electrophile (or Lewis acid). If we use a common alkyl halide, such as methyl bromide, and a common solvent, ethanol, we can examine the rate at which various nucleophiles substitute the methyl carbon. Nucleophilicity is thereby related to the relative rate of substitution reactions at the halogen-bearing carbon atom of the reference alkyl halide. The most reactive nucleophiles are said to be more nucleophilic than less reactive members of the group. The nucleophilicities of some common Nu:(–) reactants vary as shown in the following chart. Nucleophilicity: CH3CO2(–) < Cl(–) < Br(–) < N3(–) < CH3O(–) < CN(–) < I(–) < SCN(–) < I(–) < CH3S(–) The reactivity range encompassed by these reagents is over 5,000 fold, thiolate being the most reactive. Note that by using methyl bromide as the reference substrate, the complication of competing elimination reactions is avoided. The nucleophiles used in this study were all anions, but this is not a necessary requirement for these substitution reactions. Indeed reactions 6 & 7, presented at the beginning of this section, are examples of neutral nucleophiles participating in substitution reactions. The cumulative results of studies of this kind has led to useful empirical rules pertaining to nucleophilicity: (i) For a given element, negatively charged species are more nucleophilic (and basic) than are equivalent neutral species. (ii) For a given period of the periodic table, nucleophilicity (and basicity) decreases on moving from left to right. (iii) For a given group of the periodic table, nucleophilicity increases from top to bottom (i.e. with increasing size), although there is a solvent dependence due to hydrogen bonding. Basicity varies in the opposite manner. 22 2. Solvent Effects Solvation of nucleophilic anions markedly influences their reactivity. The nucleophilicities cited above were obtained from reactions in methanol solution. Polar, protic solvents such as water and alcohols solvate anions by hydrogen bonding interactions, as shown in the diagram on the right. These solvated species are more stable and less reactive than the unsolvated "naked" anions. Polar, aprotic solvents such as DMSO (dimethyl sulfoxide), DMF (dimethylformamide) and acetonitrile do not solvate anions nearly as well as methanol, but provide good solvation of the accompanying cations. Consequently, most of the nucleophiles discussed here react more rapidly in solutions prepared from these solvents. These solvent effects are more pronounced for small basic anions than for large weakly basic anions. Thus, for reaction in DMSO solution we observe the following reactivity order: Nucleophilicity: I(–) < SCN(–) < Br(–) < Cl(–) ≈ N3(–) < CH3CO2 (–) < CN(–) ≈ CH3S(–) < CH3O(–) Note that this order is roughly the order of increasing basicity. 3. The Alkyl Moiety Some of the most important information concerning nucleophilic substitution and elimination reactions of alkyl halides has come from studies in which the structure of the alkyl group has been varied. If we examine a series of alkyl bromide substitution reactions with the strong nucleophile thiocyanide (SCN) in ethanol solvent, we find large decreases in the rates of reaction as alkyl substitution of the alpha-carbon increases. Methyl bromide reacts 20 to 30 times faster than simple 1º-alkyl bromides, which in turn react about 20 times faster than simple 2º-alkyl bromides, and 3º-alkyl bromides are essentially unreactive or undergo elimination reactions. Furthermore, βalkyl substitution also decreases the rate of substitution, as witnessed by the failure of neopentyl bromide, (CH3)3CCH2-Br (a 1º-bromide), to react. 23 Alkyl halides in which the alpha-carbon is a chiral center provide additional information about these nucleophilic substitution reactions. Other investigations have shown this to be generally true for reactions carried out in non-polar organic solvents, the reaction of (S)-2-iodobutane with sodium azide in ethanol being just one example ( in the following equation the alpha-carbon is maroon and the azide nucleophile is blue). Inversion of configuration during nucleophilic substitution has also been confirmed for chiral 1º-halides of the type RCDH-X, where the chirality is due to isotopic substitution. (S)-CH3CHICH2CH3 + NaN3 ——> (R)-CH3CHN3CH2CH3 + NaI We can now piece together a plausible picture of how nucleophilic substitution reactions of 1º and 2º-alkyl halides take place. The nucleophile must approach the electrophilic alpha-carbon atom from the side opposite the halogen. As a covalent bond begins to form between the nucleophile and the carbon, the carbon halogen bond weakens and stretches, the halogen atom eventually leaving as an anion. The diagram on the right shows this process for an anionic nucleophile. We call this description the SN2 mechanism, where S stands for Substitution, N stands for Nucleophilic and 2 stands for bimolecular (defined below). In the SN2 transition state the alpha-carbon is hybridized sp2 with the partial bonds to the nucleophile and the halogen having largely p-character. Both the nucleophile and the halogen bear a partial negative charge, the full charge being transferred to the halogen in the products. The consequence of rear-side bonding by the nucleophile is an inversion of configuration about the alpha-carbon. Neutral nucleophiles react by a similar mechanism, but the charge distribution in the transition state is very different. This mechanistic model explains many aspects of the reaction. First, it accounts for the fact that different nucleophilic reagents react at very different rates, even with the same alkyl halide. Since the transition state has a partial bond from the alpha-carbon to the nucleophile, variations in these bond strengths will clearly affect the activation energy, ΔE‡, of the reaction and therefore its rate. Second, the rear-side approach of the nucleophile to the alpha-carbon will be subject to hindrance by neighboring alkyl 24 substituents, both on the alpha and the beta-carbons. The following models clearly show this "steric hindrance" effect. The two models displayed below start as methyl bromide, on the left, and ethyl bromide, on the right. These may be replaced by isopropyl, tert-butyl, neopentyl, and benzyl bromide models by pressing the appropriate buttons. (note that when first activated, this display may require clicking twice on the selected button.) In each picture the nucleophile is designated by a large violet sphere, located 3.75 Angstroms from the alpha-carbon atom (colored a dark gray), and located exactly opposite to the bromine (colored red-brown). This represents a point on the trajectory the nucleophile must follow if it is to bond to the back-side of the carbon atom, displacing bromide anion from the front face. With the exception of methyl and benzyl, the other alkyl groups present a steric hindrance to the back-side approach of the nucleophile, which increases with substitution alpha and beta to the bromine. The hydrogen (and carbon) atoms that hinder the nucleophile's approach are colored a light red. The magnitude of this steric hindrance may be seen by moving the models about in the usual way, and is clearly greatest for tert-butyl and neopentyl, the two compounds that fail to give substitution reactions. S 2 Reactions N Nucleophilic substitution reactions of alkyl halides take place by a continuum of mechanisms, defined by SN2 behavior at one extreme and SN1 behavior at the other. The essential characteristics of the SN2 process include inversion of configuration at the reaction site, sensitivity to steric hindrance in the alkyl group, and second order kinetic behavior. The orbital interactions that take place in a successful SN2 reaction are shown in the following diagram. The bonding and antibonding sigma molecular orbitals composing the C-X bond are drawn in the yellow box at the top of the diagram. As a nucleophile approaches the rear side of the carbon, the orbital containing the non-bonded electron pair (light blue) begins to overlap with the empty antibonding C-X sigma orbital (pink colored), shown on the left of the bottom line. As electrons occupy this antibonding orbital, the C-X bond is weakened. In the transition state partial bonds exists between the carbon and both the nucleophile and the halogen (the carbon orbital is essentially sp2 and is colored orange here). Finally, a full C-Nu sigma bond develops and the halogen leaves as an anion. 25 Other substitution reactions also proceed by the SN2 pathway, as illustrated by the stereoisomeric cyclohexyl tosylates in the following diagram. The bulky tert-butyl group on C4 serves to lock the chair conformation in the configuration shown, so one isomer has an equatorial tosylate function, and the other an axial tosylate. On reaction with sodium thiophenolate, both isomers undergo bimolecular substitution with inversion, the axial isomer reacting about thirty times faster than its equatorial epimer. Steric hindrance to rear side nucleophile approach by the red colored hydrogen atoms is present for each isomer, but the relief of steric crowding of the axial tosylate leaving group helps to facilitate substitution of the that isomer. SN2 reactions may be intermolecular, as in these examples, or intramolecular. By clicking on the above equations, an example of two sequential substitutions, the first intermolecular and the second intramolecular, will be displayed. Note that the intramolecular substitution shown here proceeds via the same orbital alignment described above. The reacting moieties in intramolecular reactions are incorporated in the same molecule; consequently, such reactions exhibit first order kinetic behavior instead of the usual second order kinetics. Because enolate alkylation reactions are irreversible, the strained four-membered ring product is stable under the reaction conditions. 26 In general, intramolecular forms of bimolecular reactions are faster than their intermolecular counterparts, since the reactive sites are held close together (their relative concentrations are as high as possible). The rapid lactonization of gamma and delta-hydroxy acids compared with corresponding intermolecular esterifications is another example of this principle, which is associated with a favorable entropy change. Note, however, that ring size has a profound influence on the intra- vs. intermolecular dichotomy. Maximum overlap of the electron orbital of the nucleophile with the antibonding sigma orbital of the carbon substituent requires an approach from the rear side of the carbon, ideally 180º from the leaving group. The importance of this orientation, which is shown in previous diagrams, was made clear by an experiment conducted by A. Eschenmoser over 25 years ago. The results of Eschenmoser's experiment are presented in the following illustration. Initially, two equations are written. The first describes the intermolecular methylation of a sulfone anion by methyl tosylate, a typical SN2 reaction. The second is a very similar reaction which many would consider to be intramolecular, since a methyl sulfonate moiety is positioned ortho to the sulfone function. 4. Molecularity If a chemical reaction proceeds by more than one step or stage, its overall velocity or rate is limited by the slowest step, the rate-determining step. This "bottleneck concept" has analogies in everyday life. For example, if a crowd is leaving a theater through a single exit door, the time it takes to empty the building is a function of the number of people who can move through the door per second. Once a group gathers at the door, the speed at which other people leave their seats and move along the aisles has no influence on the overall exit rate. When we describe the mechanism of a chemical reaction, it is important to identify the rate-determining step and to determine its "molecularity". The molecularity of a reaction is defined as the number of molecules or ions that participate in the rate determinining step. A mechanism in which two reacting species combine in the transition state of the rate-determining step is called bimolecular. If a single species makes up the transition state, the reaction would be called unimolecular. The relatively improbable case of three independent species coming together in the transition state would be called termolecular. 27 5. Kinetics One way of investigating the molecularity of a given reaction is to measure changes in the rate at which products are formed or reactants are lost, as reactant concentrations are varied in a systematic fashion. This sort of study is called kinetics, and the goal is to write an equation that correlates the observed results. Such an equation is termed a kinetic expression, and for a reaction of the type: A + B –––> C + D it takes the form: Reaction Rate = k[A] n[B] m, where the rate constant k is a proportionality constant that reflects the nature of the reaction, [A] is the concentration of reactant A, [B] is the concentration of reactant B, and n & m are exponential numbers used to fit the rate equation to the experimental data. Chemists refer to the sum n + m as the kinetic order of a reaction. In a simple bimolecular reaction n & m would both be 1, and the reaction would be termed second order, supporting a mechanism in which a molecule of reactant A and one of B are incorporated in the transition state of the rate-determining step. A bimolecular reaction in which two molecules of reactant A (and no B) are present in the transition state would be expected to give a kinetic equation in which n=2 and m=0 (also second order). The kinetic expressions found for the reactions shown at the beginning of this section are written in blue in the following equations. Each different reaction has its own distinct rate constant, k#. It should be recognized and remembered that the molecularity of a reaction is a theoretical term referring to a specific mechanism. On the other hand, the kinetic order of a reaction is an experimentally derived number. In ideal situations these two should be the same, and in most of the above reactions this is so. Reaction 7 above is clearly different from the other cases reported here. It not only shows first order kinetics (only the alkyl halide concentration influences the rate), but the chiral 3º-alkyl bromide reactant undergoes substitution by the modest nucleophile water with extensive racemization. Note that the acetonitrile cosolvent does not function as a nucleophile. It serves only to provide a homogeneous solution, since the alkyl halide is relatively insoluble in pure water. One of the challenges faced by early workers in this field was to explain these and other differences in a rational manner. 28 1. The S 2 Mechanism N As described in the previous section, a majority of the reactions thus far described appear to proceed by a common single-step mechanism. This mechanism is referred to as the SN2 mechanism, where S stands for Substitution, N stands for Nucleophilic and 2 stands for bimolecular. Other features of the SN2 mechanism are inversion at the alpha-carbon, increased reactivity with increasing nucleophilicity of the nucleophilic reagent and steric hindrance to rear-side bonding, especially in tertiary and neopentyl halides. Although reaction 3 exhibits second order kinetics, it is an elimination reaction and must therefore proceed by a very different mechanism, which will be described later. 2. The S 1 Mechanism N Reaction 7, shown at the end of the previous section, is clearly different from the other cases we have examined. It not only shows first order kinetics, but the chiral 3º-alkyl bromide reactant undergoes substitution by the modest nucleophile water with extensive racemization. In all of these features this reaction fails to meet the characteristics of the S N2 mechanism. A similar example is found in the hydrolysis of tert-butyl chloride, shown below. Note that the initial substitution product in this reaction is actually a hydronium ion, which rapidly transfers a proton to the chloride anion. This second acid-base proton transfer is often omitted in writing the overall equation, as in the case of reaction 7 above. (CH3)3C-Cl + H2O ——> (CH3)3C-OH2(+) + Cl(–) ——> (CH3)3C-OH + HCl Although the hydrolysis of tert-butyl chloride, as shown above, might be interpreted as an SN2 reaction in which the high and constant concentration of solvent water does not show up in the rate equation, there is good evidence this is not the case. First, the equivalent hydrolysis of ethyl bromide is over a thousand times slower, whereas authentic SN2 reactions clearly show a large rate increase for 1º-alkyl halides. Second, a modest increase of hydroxide anion concentration has no effect on the rate of hydrolysis of tert-butyl chloride, despite the much greater nucleophilicity of hydroxide anion compared with water. The first order kinetics of these reactions suggests a two-step mechanism in which rate-determining step consists of the the ionization of the alkyl halide, as shown in the 29 diagram on the right. In this mechanism, a carbocation is formed as a high-energy intermediate, and this species bonds immediately to nearby nucleophiles. If the nucleophile is a neutral molecule, the initial product is an "onium" cation, as drawn above for t-butyl chloride, and presumed in the energy diagram. PROSEDUR : I. Sintesis 2-kloro-2-metilpropana 1. Tujuan : Mengenal reaksi halogenasi melalui mekanisme reaksi substitusi nukleofilik Sintesis 2-kloro-2-metilpropana 2. Reaksi : CH3 CH3 H3C C CH3 + HCl H3C OH CH3 + H2O Cl 3. Alat : corong pisah labu Erlenmeyer 4. Bahan : HCl pekat 2-metil-2-propanol Larutan NaHCO3 5% MgSO4 anhidrat 5. Cara kerja : Ke dalam corong pisah 250 mL, masukkan 75 mL HCl pekat yang telah didinginkan sekitar 10°C Tambahkan 22 gr 2-metil-2-propanol Kocok secara periodik selama 20 menit dan keluarkan gas yang terbentuk 30 Pisahkan fasa air dan segera cuci fasa organik dengan 25 ml larutan NaHCO3 5% (keluarkan gas yang terbentuk) dan kemudian cuci dengan air Selanjutnya fasa organik yang telah dicuci dikeringkan dengan MgSO4 anhidrat. Saring II. Pembuatan 2-bromo butana 1. Tujuan : Sintesis 2-bromo butana Mempelajari reaksi substitusi nukleofilik 2. Reaksi : H3C H C H2 C CH3 + HBr OH 3. Alat H2 C H3C CH3 + H2O Br : seperangkat alat destilasi sederhana labu alas bulat 250 mL labu Erlenmeyer corong pisah 4. Bahan : 2-butanol MgSO4 / Na2SO4 anhidrat HBr pekat batu didih HCl pekat larutan NaHCO3 5% 5. Cara kerja : Ke dalam labu alas bulat 250 mL masukkan 24 gr 2-butanol, 150 mL HBr pekat dan batu didih Gabungkan labu alas bulat dengan seperangkat alat distilasi sederhana 31 Lakukan distilasi selama ± 2 jam sampai volume dalam labu tinggal separuhnya Tuang distilat yang diperoleh ke dalam corong pisah Pisahkan dua fasa yang terbentuk Cuci fasa organik tersebut dengan 25 mL HCl pekat, kemudian dengan larutan NaHCO3 5% dan dengan air Ambil fasa organik dan keringkan menggunakan MgSO 4 / Na2SO4 anhidrat Gambar 10. Reaksi subtitusi menggunakan distilasi sederhana 32