Word file (31 KB )
advertisement
")
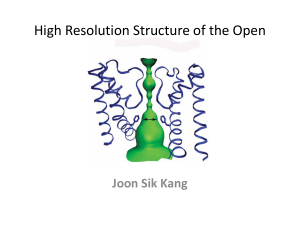
Supplementary Information for Sukharev et al. Description of the Molecular Modeling Process and Rationale In general, models of all conformations were developed to be consistent with the experimental data and to satisfy a series of modeling criteria 17 related to (1) energetically favorable interactions of residues with water, lipids, and each other; (2) favorable backbone and side chain conformations; (3) clustering of highly conserved residues and exposure of poorly conserved residues along the series of conformations representing the transition 18; (4) helix packing theory 11,19 ; and (5) the thickness of a lipid bilayer and formation of a tightly packed protein barrier between membrane lipids and water of the pore. Specifically, the following procedures were used to develop the structural models. First, models of the helical portions of EcoMscL were developed using a program that generates helices with idealized backbone conformations and side chains in the conformation that occurs for that residue most frequently in helices. The M1 and M2 helices were then matched to those of the TbMscL crystal structure. The S1 helices and S3 helices were arranged to generate two different bundles of helices with 5-fold symmetry that each have a highly conserved hydrophobic core and in which helices packing is consistent with ‘knobsinto-holes’ and or ‘ridges-into-grooves’ helix packing theories. The initial models of the S1 and S3 bundles were left-handed, which allows the helices to pack in a manner typical of coiled-coils. These were placed in the cytoplasmic region with the S3 bundle located as it is in the TbMscL crystal structure and with the S1 bundle positioned co-axially between the S3 and M1 bundles. Segments connecting the helices were then constructed manually choosing backbone and side chain conformations that are energetically favorable. The axial location and radial orientation of the S1and S3 bundles were adjusted to enhance energetically favorable interactions and conformations of the linking segments. The periplasmic loop of EcoMscL was so unlike that of TbMscL that it was generated strictly to satisfy our criteria, using an helix for the initial region and a distorted hairpin for the remaining portion. Some side chain conformations were then adjusted to alternative conformations that are energetically favorable to eliminate energetically unfavorable contacts and to enhance energetically favorable interactions such as salt bridges and hydrogen bonds. The energy of the model was then minimized using CHARMM with the structure constrained to maintain five-fold symmetry about the pore’s axis. The minimized model was then examined visually and adjustments were made manually to make the models more consistent with our criteria. The adjusted model was then minimized again. This process was repeated until no additional improvements could be made. Two classes of models have been developed for the open conformation, 10-helix and 5-helix pore models. The most obvious mechanism for opening the pore is one in which the M1 helices move radially outward between the M2 helices, creating an open pore lined by both M1 and M2 helices (10-helix pore) 5,6,9. We developed several models of this type. None of these models satisfied our criteria as well as the 5-helix pore models presented here. Specifically, more hydrophobic side chains, primarily on M2, were exposed to water inside the pore and there were fewer energetically favorable contacts among highly conserved residues. In the framework of 5-helix pore model, the opening of the channel is a result of a gradual expansion of the barrel associated with increased tilts of transmembrane helices. First, the tightly packed M1 and M2 pair from adjacent subunits was translated radially outward about one Å and the tilts and orientations of this pair were adjusted to eliminate gaps between the helices, to minimize the amount of hydrophobic surface that is exposed to water inside the channel, and to maintain ‘4-4 ridges-into-grooves’ packing between adjacent M1 helices. The locations and orientations of the S1 and S3 bundles and conformations of all of the connecting segments were constructed in a manner that minimized changes from the closed conformation and that maintained a tight seal between the S1 bundle and the cytoplasmic entrance to the transmembrane pore. The initial, slightly expanded, structure was then minimized using CHARMM. The process of iterative manual adjustments of this model followed by energy minimization was repeated as before. The process of expanding the transmembrane pore structure in small steps and optimizing the structure was repeated in successive steps until, when the M1 pore had a diameter of 10-15 Å, the increase of the tilts of M1 and M2 reduced the transmembrane distance spanned substantially, the exposed hydrophobic surface on M1 and M2 began to increase substantially, opening began to form between the S1 bundle and the M1M2 pore, and the M2-S3 linker became very extended. The amphipathic S3 helices were then placed parallel to the membrane at its cytoplasmic surface in a location where they blocked the openings between the S1 bundles and the pore. In these expanded-closed models the highly conserved hydrophobic residues of the S3 helices interact with lipids and/or with other highly conserved hydrophobic residues on M1 and M2, while their highly conserved charged residues form salt bridges with other highly conserved charged residues of the S1 helices. The process of gradually expanding the transmembrane pore while maintaining the tightly packed S1 bundle was then continued until its diameter was ~25 Å and the R13-G14-N15 segment linking S1 to M1 was fully extend. The S1 bundle was then adjusted to form a right handed bundle similar to that formed by the M1’s of the closed conformation. The hydrophobic core at the axis of this bundle is formed only by the F7 and I3 side chains. The F10 side chain is more peripheral; it fits between the I3, I4, F7, and R8 side chains of the adjacent S1 helix (see Fig. 3). The right-handed S1 bundle was further stabilized by the binding of the highly conserved E6 side chain to the N-terminus of the adjacent S1. The C-terminus of the S1’s in the right-handed bundles is farther from the axis of symmetry than in the left-handed bundles, which reduced the tension on the S1-M1 linker and allowed additional expansion of the transmembrane pore prior to disruption of the S1 bundle. When the pore reached a diameter of about ~30 Å, the S1-M1 linker became fully extended even when the S1 helices are maximally tilted for the right-handed bundle. This was judged to be the closed conformation with the largest possible transmembrane pore. Models were then developed of open conformations in which S1 helices dock on the parameter of the channel. These models were developed so that the highly conserved F7 and F10 side chains of S1’s could interact with highly conserved side chains of I25, A28, and F29 of M1 and F85, A89, and F93 of M2. This docking displaces the S3 helix from its previous site. S3 was repositioned parallel to the S1 helix so that its hydrophobic residues should interact with lipid alkyl chains at the cytoplasmic surface while it’s highly conserved charged residues still form salt bridges with highly conserved charged residues of S1. This type of model was expanded a bit more until the pore’s diameter of was ~38Å, which we deemed to be the largest pore that could be developed realistically with this type of model. This size is consistent with experimental findings 7,8. Also, during the expansion process, portions of the S2 periplasmic loop were gradually pulled into the transmembrane region. The loop was constructed so that most of its hydrophobic residues are buried when the channel is closed but become exposed to lipid alkyl chains on outer surface as the channel expands. In all, we developed models with transmembrane pores having thirteen different sizes, ranging from fully closed as in the crystal structure to fully open with a diameter of 38 Å. The transitions involved little changes in secondary structures with most of the conformational changes occurring at or near glycines or prolines (G14 of the S1-M1 linker; P43 and P44 between the M1 and S3 helix; G50 and G51 between the S3 helix and first S3 β strand; G66 and P69 in the β turn region of S3; G76 between S3 and M2; and P109, P113, and P115 of the linker between the M2 and S3 helices). Glycines and prolines tend to introduce flexibility into protein structures. With the exceptions of the break down of the S1 and S3 bundles and docking of the S1 and S3 helices on the perimeter of the channel, all of the conformational changes between successive steps are relatively small with no apparent steric energy barriers for the transitions from one step to the next. In all conformations, almost all of the hydrophobic side chains are either buried, partially buried, or located where they should be exposed to lipid alkyl chains. In the open conformation hydrophobic side chains of M1, V23, F29, V33, V37, and I41, are partially exposed to water in the pore, which should contribute to the instability of the open conformation when the membrane is not stretched. All of the hydrophilic side chain atoms are exposed to water and/or form energetically favorable salt bridges or hydrogen bonds in all conformations. The M1 helices have a series of small ambivalent glycine, alanine, and serine residues that are buried in the closed conformation and become exposed in the expanded conformations. The energy change in moving these residues from a buried protein environment to an exposed aqueous environment is small. The barrier between the hydrophobic lipid phase and interior of the pore is solid in all conformations and the length of the hydrophobic surface on the exterior of the model is sufficient to span a hydrophobic region that is ~30 Å thick. Sequences of thirty five MscL homologs were aligned and the degree of conservation at each position of alignment was determined. With the exception of some glycine and proline residues that affect the backbone structure, all highly conserved residues interact with other highly conserved residues in at least one conformation, and most interact with other highly conserved residues in all conformation. These highly conserved interactions are energetically favorable; e.g., all charged side chains form salt bridges in at least one conformation, all hydrophobic side chains interact with other hydrophobic side chains, and aromatic residues tend to form clusters.