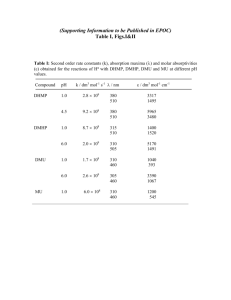
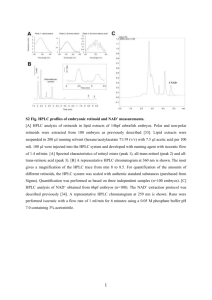
(400 MHz) spectroscopic data of orientin and vitexin
advertisement
 spectroscopic data of orientin and vitexin")
SUPPLEMENTARY MATERIAL Isolation and purification of orientin and vitexin from Trollius chinensis Bunge by high-speed counter-current chromatography Xiao-xue Yua,b, Jie-yun Huanga,b, Dan Xuc, Zhi-yong Xiea,b , Zhi-sheng Xiea,b and Xin-jun Xua,b a School of Pharmaceutical Sciences, Sun Yat-Sen University, 510006 Guangzhou, P. R. China b Guangdong Technology Research Centre for Advanced Chinese Medicine, 510006 Guangzhou, P. R. China c Guangdong Union Biotechnology Co., Ltd, 510663 Guangzhou, P. R. China Correspondence: Xinjun Xu, School of Pharmaceutical Sciences, Sun Yat-Sen University. 132 Waihuan Rd. East, Guangzhou Higher Education Mega Center, Guangzhou, 510006, P. R. China. Tel: +86 13924061779, +86 20 39943041. E-mail address: xxj2702@sina.com Orientin and vitexin are the two main bioactive compounds in Trollius chinensis Bunge. In this study, a rapid method was established for the isolation and purification of orientin and vitexin from Trollius chinensis Bunge using high-speed counter-current chromatography in one step, with a solvent system of ethyl acetate-ethanol-water (4:1:5, v/v/v). A total of 9.8 mg orientin and 2.1 mg vitexin were obtained from 100 mg of the ethyl acetate extract, with the purities of 99.2% and 96.0%, respectively. Their structures were identified by UV, MS, and NMR. The method was efficient and convenient, which could be used for the preparative separation of orientin and vitexin from Trollius chinensis Bunge. Experimental Apparatus The HSCCC instrument was QuikPrepTM Chassis Mk5 high-speed counter-current chromatography (AECS-QuikPrep Ltd, Bristol, UK). The unit had four identical coils of 3.2 mm od and 2.16 mm id, and the applicable rotational speed was 860rpm. Each coil had a volume of approximately 120 mL. Each coil in this configuration had a beta range of 0.85 to 0.62, but in this paper, only one coil was used for separation. This Quattro CCC system was equipped with a series II HPLC pump (SSI, USA), a sample injection valve with a 10 mL sample loop and an SPD-10AVP detector (SHIMADZU, Japan). The work station was N2000 (Zhejiang University, China). Other equipments included a Lab alliance HPLC with a 1500 pump, an AS1000 autosampler, and a UV6000 detector (SSI, USA), a Finnigan LCQ DECA XP Liquid Chromatography Mass Spectrometer (Thermo, USA), an SB25-12DTD ultrasound machine (Xinzhi Biotechnical. Ltd, Ningbo, China), an electronic balance (KERN ABT 220-5DM, 0.1mg, Germany), an RE-3000 rotational vacuum concentrator (Shanghai Yarong, China), and a Bruker Avance Ⅲ 400 Nuclear Magnetic Resonance Spectrometer (Germany). Reagents and materials The flowers of Trollius chinensis Bunge were purchased from Guangdong Guokang Pharmaceutical Ltd. (Batch Number: 20120207) and authenticated by Associate Professor Xu (Lab of Pharmaceutical Analysis and Quality Assessment, Sun Yat-sen University). Acetonitrile of HPLC grade was purchased from SK Chemicals (Korea). All organic solvents used for HSCCC separation were of analytical grade and purchased from Tianjin Damao Chemical Reagent Factory (Tianjin, China). Ultrapure water was obtained from a Milli-QRG purification unit (Millipore, Bedford, MA, USA). Dimethyl sulfoxide (DMSO-d6) used for NMR determination was made by Cambridge Isotope Laboratories Inc. D101 macroporous resin was purchased from Cangzhou Bon Adsorber Technology Co. Ltd (Cangzhou, China). Preparation of crude sample About 100 g dried flowers of Trollius chinensis Bunge were pulverized and extracted for three times (30 min for each time) with 1000 mL of 60% ethanol aqueous each time. After being filtered, the extract was evaporated to dryness at 40 °C by rotary vaporization under reduced pressure, and then the ethanol extract was dissolved in water and added into a glass column (30 mm×200 mm, contained about 100 g D101 macroporous resin). Water was first used to remove some water soluble components and then 30% ethanol aqueous was used for elution. The effluent was collected and evaporated to dryness by rotary vaporization. Then, the dried eluate was suspended in water and extracted three times with equal volumes of ethyl acetate. The ethyl acetate layers were concentrated under vacuum to dryness at 40 °C. Measurement of partition coefficient The K-values were determined by HPLC as follows: a suitable amount of crude sample was added to 2.0 mL of the lower phase in the test tube and analysed by HPLC where the peak area of the lower phase was recorded as A1. Then 2.0 mL of the upper phase was added to the lower phase and was shaken vigorously, and the lower phase was analysed by HPLC again after the equilibration was established. The peak area of the lower phase was recorded as A2. The K-value was calculated according to the following equation: K = CU/CL = (A1- A2)/ A2. Preparation of two-phase solvent system and sample solution The ethyl acetate-ethanol-water (4:1:5, v/v/v) system was shaken vigorously in a separatory funnel and equilibrated at room temperature overnight. Then, the upper phase and the lower phase were separated and degassed by sonication for 30 min before use. The upper phase was used as the stationary phase and the lower phase was used as the mobile phase. The sample solution was prepared by dissolving the 100 mg crude sample in 10 mL solvent mixture consisting of equal volumes of each phase of the two-phase solvent system used for HSCCC separation. HSCCC separation procedure The coiled column was firstly filled with the upper phase of ethyl acetate-ethanol-water (4:1:5, v/v/v) at a flow rate of 8 mL·min-1. The rotation speed was set at 860 rpm before the lower phase was pumped into the column from the head to tail at a flow rate of 2 mL·min-1. The sample solution was loaded into the injection valve after the system reached hydrodynamic equilibrium. The effluent from the column was monitored at the wavelength of 254 nm and collected into test tubes with a fraction collector set at 3 min for each tube. The fractions were analysed by HPLC later. All fractions of the same pure compound were combined and evaporated under reduced pressure. HPLC analyses and identification of HSCCC peak fractions A Grace C18 column (150 mm × 4.6 mm, 5 μm) with a C18 guard column (10 mm × 4.6 mm, 5 μm) was used. The mobile phase consisted of aqueous acetic acid (1%, v/v) (A) and acetonitrile (B). The system was run with a gradient program at the flow rate of 1 mL·min-1: 0~40 min: 15% B → 40% B, 40~50 min: 15% B. The sample injection volume was 10 μL. The wavelength was set at 254 nm and the column temperature was 35 °C. The identification of HSCCC fractions was carried out by UV, MS, 1H NMR, and 13C NMR. Figure Captions Figure S1. HPLC chromatogram of crude extract of Trollius chinensis Bunge. Figure S2. HPLC chromatogram of crude extract of Trollius chinensis Bunge after pretreatment by D101 macroporous resin and extracting by ethyl acetate. Figure S3. HSCCC chromatogram of Trollius chinensis Bunge using No.4 as solvent system. Figure S4. HSCCC chromatogram of the extract of Trollius chinensis Bunge using No.5 as solvent system. Figure S5. HPLC chromatograms of compound 1 (A), compound 2 (B). Figure S1. HPLC chromatogram of crude extract of Trollius chinensis Bunge. Figure S2. HPLC chromatogram of crude extract of Trollius chinensis Bunge after pretreatment by D101 macroporous resin and extracting by ethyl acetate. Figure S3. HSCCC chromatogram of Trollius chinensis Bunge using No.4 as solvent system. Figure S4. HSCCC chromatogram of the extract of Trollius chinensis Bunge using No.5 as solvent system. Figure S5. HPLC chromatograms of compound 1 (A), compound 2 (B). TABLE S1 The K-values of the target components measured in different solvent systems No. Solvent system Ratio(v/v/v/v or v/v/v) K1 K2 1 n-hexane-ethyl acetate-methanol-water 1:5:1:5 0.16 0.52 2 Ethyl acetate-n-butanol-water 4:1:5 2.68 5.31 3 Ethyl acetate-n-butanol-water 2:1:3 3.77 5.72 4 Ethyl acetate-methanol-water 4:1:5 0.58 1.30 5 Ethyl acetate-ethanol-water 4:1:5 0.99 1.70 6 Ethyl acetate-acetone-water 4:1:5 0.36 0.81 *K, solute concentration in the upper mobile phase divided by that in the lower stationary phase. K1, the K-value of compound 1; K2, the K-value of compound 2. TABLE S2 1H NMR (400 MHz) and 13C NMR (100 MHz) spectroscopic data of orientin and vitexin (DMSO-d6, δ in ppm). compound 1 Position δC δH multiplicity(J in compound 2 Position δC Hz) 2 164.11 3 102.42 4 182.03 5 160.41 6 98.17 7 δH multiplicity(J in Hz) 2 163.37 3 102.14 4 181.53 13.17 s 5 161.28 13.14 s 6.27 s 6 99.01 6.16 s 162.66 7 161.28 8 104.57 8 104.72 9 156.03 9 156.11 10 104.03 10 103.15 1' 122.02 1' 121.58 2' 114.09 2' 128.70 7.98 d(7.6) 3' 145.84 3' 115.85 6.88 d(8.4) 4' 149.67 4' 160.41 5' 115.69 6.87 d(8.3) 5' 115.85 6.88 d(8.4) 6' 119.40 7.54 d(8.3) 6' 128.70 7.98 d(7.6) 1'' 73.42 4.69 d(9.8) 1'' 72.25 4.74 d(9.8) 2'' 70.81 3.85 d(9.1) 2'' 70.96 3.85 m 3'' 78.80 3.26 t(8.0) 3'' 78.79 3.27 m) 4'' 70.74 3.37 t(8.7) 4'' 70.56 3.35 d(8.5) 5'' 82.01 3.26 t(8.0) 5'' 81.71 3.27 m 6'' 61.68 6'' 61.31 6.65 s 7.49 brs 3.80 d(12.3) 3.55 dd(11.5, 6.2) 6.68 s 3.75 d(11.2) 3.53 m