mec12911-sup-0002-AppendixS2
advertisement

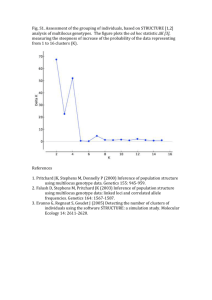
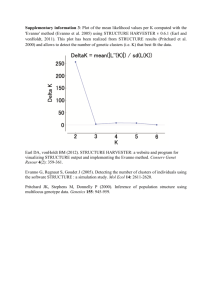
Medrano, Herrera and Bazaga Appendix S2 – 1 Appendix S2. Genetic structure of Helleborus foetidus plants sampled for this study Methods To examine whether the 200 H. foetidus plants sampled for this study from ten locations were genetically structured, and to define the most likely number of genetic clusters (K) represented in the sample, we used Bayesian clustering analyses as implemented in STRUCTURE v2.3 (Pritchard et al. 2000; Falush et al. 2003, 2007). These analyses were performed on the data set of 270 AFLP fragments, using the following parameters: length of burn-in period = 500,000, number of Markov chain Monte Carlo reps after burn-in = 1,000,000, admixture model, and correlated allele frequencies. To test the stability of the results, 30 iterations per K level were performed from K = 1 through K = 10. The most probable number of genetic clusters was determined by calculating the ad hoc statistic delta K, which identifies the highest rate of change in the log-likelihood between successive Ks (Evanno et al. 2005), in STRUCTURE HARVESTER (Earl & vonHoldt 2012). The program CLUMPP (Jakobsson & Rosenberg 2007) was used to compile individual assignments across all 30 replicates for the most likely K, with the Greedy algorithm and 5000 reps. Individual group membership probabilities for individual plants were plotted for visualization using DISTRUCT 1.1 (Rosenberg 2004). Results The results of the STRUCTURE analyses revealed that the recognition of two clusters (K = 2) best reflected the genetic structure present in our data (Fig. S2A). At K = 2, a southern group of four populations (PLL, SCA, CFU, and ESP respectively) were clearly differentiated from the two northernmost populations (TEJ, CAN) as well as the four geographically intermediate populations (NAV, MES, FBE, and VCU; Fig. S2B). The vast majority of individuals (86%) were assigned to one of these two genetic clusters with a probability of 0.9 or more. Some admixture between these two main lineages occurred in three of the four central populations (MES, NAV, and FBE) as denoted by a certain proportion of admixed individuals (Fig. S2B). Medrano, Herrera and Bazaga Appendix S2 – 2 Figure S2. Results of the Bayesian clustering analysis using STRUCTURE of the 200 plants x 270 AFLP marker data matrix. (A) Estimation of the most probable number of genetic clusters present in the sample (K) for K=1-10 and 30 independent STRUCTURE simulations: posterior probabilities of the data LnP(D) (open circles) and values of Evanno’s Delta K (filled circles). (B) Maps showing the location of the ten studied populations in south eastern Europe (left panel) and the distribution of the two different genetic groups obtained using STRUCTURE (right panel) represented as: the proportion of the two genetic pools detected in each population (pie charts); and the probability of membership to the two inferred clusters for each of the 200 Helleborus foetidus individuals sampled (horizontal lines inside the barplot). Colors represent the estimated probabilities of membership to each cluster. Population codes as in Appendix S1. A) B) Medrano, Herrera and Bazaga Appendix S2 – 3 References Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4, 359–361. Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology, 14, 2611–2620. Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics, 164, 1567–1587. Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Molecular Ecology Notes, 7, 574–578. Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics, 23, 1801–1806. Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics, 155, 945–959 Rosenberg NA (2004) Distruct: a program for the graphical display of population structure. Molecular Ecology Notes, 4, 137–138.