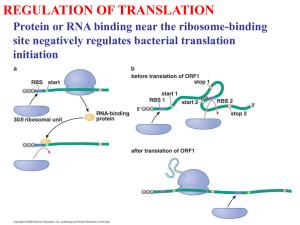
Cotranslational Protein Folding (add more later)
advertisement
")
Cotranslational Protein Folding Kinetics and the Degeneracy of the Genetic Code Jill Simard, Professor Crane, Pomona College, March 2007 Introduction It wasn’t long after Beadle and Tatum published their “one gene, one enzyme” hypothesis in 1941 that exceptions began to emerge (1). In particular, it was discovered that RNA splicing and post-translational protein modifications can lead to different proteins from a single transcribed sequence. However, until recently it was widely held that a single mRNA encoding a specific amino acid sequence will be translated to give identical protein products, assuming that the co- and post-translational modifications are the same. That is, it was assumed that a protein’s overall structure and consequent function was dictated solely by its amino acid sequence. This became known as Anfinsen’s principle (2). While a large number of proteins continue to support Anfinsen’s principle, in recent years we have also seen cases in which varying rates of translation can affect protein cotranslational folding and therefore protein structure and behavior, even if the primary sequences are held constant. Varying rates of translation in vivo can be attributed to a wide range of factors, such as variations in tRNA and transcription factor concentrations, as well as changes in codon usage (3). This last factor is particularly interesting and will be the primary focus of this paper; it suggests that codons previously thought to be synonymous can in fact have different effects on protein conformation. Thus, “silent” mutations may not necessarily be truly silent, and the degeneracy of the genetic code is more relevant to biological function than was previously thought. This recent revelation has profound consequences both for experimental protein expression and for the protein misfolding that has been shown to lead to a wide range of conformational diseases. Genetic code degeneracy (a review) and single nucleotide polymorphisms (SNP’s) Because each codon contains three nucleotides and there are four possible nucleotides, sixty-four different codons are possible. There are only twenty different amino acids to encode, so in most cases multiple codons can encode a single amino acid. As seen in Figure 1, the codons corresponding to a certain amino acid frequently differ by only one nucleotide, making it possible for single nucleotide polymorphisms (SNPs) or synonymous nucleotide substitutions to occur without changing the primary structure of a protein. Cases involving a synonymous single nucleotide substitution are often called “silent mutations,” and one popular explanation for the degeneracy of the genetic code has been that such “silent” mutations decrease the prevalence of amino acid sequence alterations, which could ultimately affect protein efficacy (4). Figure 1. The genetic code and its degeneracy. Cotranslational Protein Folding and Kinetics Codon Usage It has been shown that E. Coli and other organisms exhibit strong codon biases; for example, if there exist four possible codons for a single amino acid, often one of those codons will be present with significantly greater frequency. It has also been seen that concentrations of cognate tRNAs are directly proportional to codon usage frequency (5). Thus, it is often the case that the more rare the codon, the more slowly it is translated. Furthermore, researchers have found a tendency for genes to contain clusters (of around ten or less codons) of either frequent or rare codons (6), and the rare codon clusters tend to encode turns, loops, and domain linkers within the translated protein (7). These studies, along with supporting research, have lead to the now widely-held belief that the genetic code’s degeneracy affects cotranslational protein folding by controlling the rate of translation at certain regions. Such controlled translational rates can separate the partial folding events that occur during cotranslational protein folding, which make certain conformations in the final protein more probable. Experimental support that codon changes can lead to changes in protein structure, behavior, and function. In the past decade a significant number of cases have been uncovered in which the use of a non-wild-type synonymous codon (which would not change the protein’s primary structure) alters the resulting protein’s normal folding. In many cases this altered folding as been shown to also result in altered protein behavior and function. Studies have been conducted on both prokaryotic and eukaryotic systems. One recent study was performed using the EgPABP1 gene, which was expressed and studied in E. coli. The resulting protein contains a short region of five codons connecting two α helices, as indicated in Figure 2. In the wild-type, the five codons present are rare; the researchers changed certain nucleotides to make seven variant sequences of five frequent codons that were synonymous to the originals. They then employed an in vivo stress response-induced protein-folding reporter to examine any misfolding of the mutants, and also analyzed differences in solubility using SDS-PAGE and Western blotting. Figure 2. Two views of the EgPABP protein (rotated 90°). The arrows correspond to a turn region between two α helices (amino acids 22-26). This region contains rare codons in the wild-type; frequent codons were substituted. In one of the seven variants (variant 6, sequence shown in Table 1), both decreased solubility and aggregation were indicated, while it was confirmed that the variant in fact had the same amino acid sequence as the wild-type. The researchers hypothesize that in this particular variant, the accelerated translation rate altered the initial folding steps of the protein such that post-translational protein folding was also perturbed. The misfolding presumably exposed more hydrophobic residues, leading to a change in solubility (8). It is unknown what factors in particular lead to a marked difference in variant 6’s folding, while the behavior of the other variants remained unchanged. Table 1. Nucleotide sequences in the EgFABP1 wild-type and variants. Variant Wild type 1 2 3 4 5 6 7 Sequence F27 E21 R22 L23 G24 V25 D26 GAA GAA GAA GAA GAA GAA GAA GAA CGC CGT CGT CGT CGT CGT CGC CGT CTT CTT CTT CTT CTT CTG CTG CTG GGG GGG GGT GGG GGG GGG GGT GGT GTG GTG GTG GTT GTG GTG GTT GTT GAT GAT GAT GAT GAC GAT GAT GAC TTC TTC TTC TTC TTC TTC TTC TTC Komar and colleagues (9) have published research similar to that of the study just described, replacing rare codons with frequent codons in a protein expressed in E. coli, but this study in addition found evidence that the silent mutations affecting translational rates also impacted protein activity. Here the CATIII (chloramphenicol aminotransferase III) gene was used, and within a certain region 16 synonymous codon substitutions were made, as shown in Figure 3A. To examine translational kinetics, the group used SDSPAGE analysis—the longer a certain mRNA region spends on the ribosome, the greater the amount of “nascent peptide chains of the sizes corresponding to location of their respective rare codons.” Therefore, the darker the band on the gel electrophoresis, the slower that region was translated. Figure 3B shows not only that the kinetics of translations in non-uniform, but also indicates that substitution with more frequent codons in the mutant resulted in faster translation. This is particularly evident between the 133-136 kDa region. By using published data on individual codons’ translation rates, the researched had predicted that their synonymous substitutions would result in a twofold decrease in told total translation rate of their gene. In fact, the phosphorimager scan data of the SDS gel (Figure 3C) indicates in 2.7-fold decreased total translation time in the mutant. Figure 3. A: Wild-type and mutant CATIII sequences. B: SDS gel electrophoresis autoradiogram of CAT nascent chains. Column 1 is the mutant and column 2 is the wild-type, both after 3 min. of translation. Molecular mass in kDa. C: Phosphorimager scan of the products shown in B. At different stages in translation, aliquots of the wild-type and the synonymous-codon mutant (run under the same conditions) were removed in order to test for activity. As Figure 4 shows, the mutant exhibits a 20% decreased activity. Figure 4. Comparison of the specific activity of chloramphenicol acetyltransferase wild-type and mutant enzymes in an E. coli S30 extract cell-free system. The results are the averages of 5 trials. Specific activity is defined as the ratio of substrate conversion to the total amount of the radioactively labeled substrate at any given time. Further evidence of codon usage’s importance in biological protein function has been published just this year (10), this time involving synonymous SNPs in the Multidrug Resistance 1 (MDR1) gene coding for P-glycoprotein, an ATP-driven efflux pump. The protein plays an important role in drug pharmacokinetics and cancer cell drug resistance, and extensive research has been conducted on its numerous polymorphisms. Using multiple cell systems, researchers have shown that an SNP in certain MDR1 haplotypes alters the substrate specificity of P-glycoprotein. Their results from FACS analysis indicate that while the transport function does not change amongst the wild-type and polymorphic P-glycoproteins, the efficacy of the P-glycoprotein inhibitors verapamil and cyclosporine A decreases when the P-glycoprotein is a polymorph containing the SNP in question. They also found that the differences in efficacy between the wild-type and the haplotypes became more pronounced as more P-glycoprotein was translated. While they have not yet experimentally explored why this occurs, it is thought that as certain tRNA’s become more depleted (as translation goes on for a longer period of time), codon usage becomes more important. Treatment of both the haplotypes and the wild-type with trypsin indicates that the altered function in the presence of inhibitors is related to a conformational change amongst the variants. The wild-type and the haplotypes differed in their susceptibility to trypsin—the haplotype required a 3.4 fold greater concentration for 50% degradation— which suggests a discrepancy in tertiary structure. As in the other experimental cases, the SNP studied involved a change between rare and frequent codons: the wild-type codon GGC, exhibits 34% usage; the SNP produces the codon GGT, which has 16% usage. This data, as before, points to the significance of translational kinetics in protein folding. There are a few other ways in which synonymous codon usage could affect protein folding that has not been thoroughly tested and supported, but are nevertheless worth mentioning. One is the role of mRNA secondary structure. SNPs change the nucleotide sequence and could therefore change the way in which the mRNA typically folds, possibly altering the way it interacts with the ribosome during translation. Another thought is that synonymous codon choice could alter structurally important interactions between the charged tRNA’s and either the ribosome or the nascent peptide (11). Relevance of the effect of the genetic code’s degeneracy on protein folding Consequences for experimental work on proteins Given that SNPs have been shown to alter protein structure and function, it would be wise to pay more attention to the nucleotide sequence used for expression of a certain protein. The environment in which the protein is translated would also seem to be important, given what has recently been discovered about codon usages, tRNA concentrations, and translation rates. Each species has its own codon usage frequency, and the tRNA concentrations between species (and therefore codon translational rates) vary accordingly. Thus, in cases of heterologous gene expression, misfolding can occur due to the fact that the codon translational rates of the host are different from those in the gene’s natural organism (3). Consequences for pharmacotherapy Kimchi-Sarfaty et al.’s work on synonymous SNPs in the MDR1 gene (10), which was discussed earlier, highlights significant variations in the resulting protein’s response to certain drugs. As the synonymous SNPs are occur naturally, it can safely be assumed that there are significant variations in how people react to certain P-glycoprotein inhibitors. As pharmacotherapy becomes increasingly personalized and tailored to an individual’s genes, the effects of synonymous SNPs should be considered. The potential role of “silent” mutations in protein misfolding and conformational diseases When a polypeptide folds into a non-functional conformation, there are several biological pathways that exist to help the polypeptide achieve the functional conformation. Competing with these pathways, however, are protein degradation and aggregation, both of which can lead to a vast number of pathologies. Aggregation of misfolded proteins has been implicated in Alzheimer’s disease, Creutzfeld-Jakob disease, and sickle cell anemia; degradation has been linked to cystic fibrosis, familial hypercholesterolemia, and a vast array of other diseases (12). Thus far many of the mutations known to cause these pathologies are missense mutations. However, given the ability of synonymous codon substitutions to affect protein folding, “silent” mutations should be considered, as well. The research in this field is recent enough that few examples are available in which synonymous single nucleotide substitutions have led to pathologies. Nevertheless, there are multiple cases in which missense and nonsense mutations have failed to account for many misfolding cases. For example, Kimchi-Sarfaty, et al., note that missense and nonsense mutations account for only 60% of the cases of pseudoxanthoma elasticum, a disease caused by a mutation in the MPR6 gene (10).