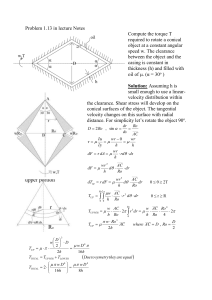
Supplementary Methods
advertisement

Supplementary methods Growth of algae Chlamydomonas reinhardtii (wild-type strain 12) was a gift from Saul Purton, University College London, UK. Cyanidium caldarium was a gift from David Vernon, University of Leeds, UK. Amphidinium operculatum and A. carterae were a gift from Chris Howe, University of Cambridge, UK. Euglena gracilis was a gift from Alan Battersby, University of Cambridge, UK. Thlalassiosira pseudonana (CMPP 1335) was received from the Provasoli-Guillard National Center for Culture of Marine Phytoplankton, Maine, USA. All other algal strains were received from the Culture Collection of Algae and Protozoa (CCAP), Oban, UK. The conditions and media used for the growth of different algal strains are summarised in the table below. With the exception of Euglena minimal medium and Cyanidium medium1, used to assess the vitamin B12 requirements of Euglena gracilis and Cyanidium caldarium respectively, the recipes for all of the other media used in this study can be found on the CCAP website (www.ife.ac.uk/CCAP/). Species Porphyridium purpureum (CCAP 1380/3) Rhodella maculata (CCAP 1388/2) Cyanidium caldarium Cyanophora paradoxa (CCAP 981/1) Chlamydomonas reinhardtii Haematococcus pluvialis (CCAP 34/6) Lobomonas rostrata (CCAP 45/2) Mantoniella squamata (CCAP 1965/1) Pyramimonas disomata (CCAP 67/8) Tetraselmis verrucosa (CCAP 66/46) Chromulina chionophila (CCAP 909/9) Poterioochromonas malhamensis (CCAP 933/1C) Thalassiosira pseudonana Skeletonema pseudocostatum (CCAP 1077/7) Hemiselmis virescens (CCAP 984/5) Rhodomonas maculata (CCAP 979/14) Pavlova gyrans (CCAP 940/1B) Pavlova sp. (CCAP 931/2) Gymmodium catenatum (CCAP 1117/7) Amphidinium operculatum Amphidinium catenatum Oxyrrhis marina (CCAP 1133/5) Euglena gracilis Medium PE Conditions 25C, shaking, continuous light F/2 + Si Troxler, 1972 JM TAP Euglena medium JM F/2+ Si ASW F/2 + Si BB JM:SE 18C, standing, 16h:8h light:dark 37C, shaking, dim light 18C, standing, 16h:8h light:dark 25C, shaking, continuous light 25C, shaking, continuous light 25C, shaking, continuous light 18C, standing, 16h:8h light:dark 18C, standing, 16h:8h light:dark 25C, shaking, continuous light 18C, standing, 16h:8h light:dark 18C, standing, 16h:8h light:dark F/2 + Si F/2 + Si 18C, standing, 16h:8h light:dark 18C, standing, 16h:8h light:dark ASW ASW F/2 + Si F/2 + Si F/2 + Si ASW or F/2 ASW F/2 + Si Euglena medium 18C, standing, 16h:8h light:dark 18C, standing, 16h:8h light:dark 18C, standing, 16h:8h light:dark 25C, shaking, continuous light 18C, standing, 16h:8h light:dark 18C, standing, 16h:8h light:dark 18C, standing, 16h:8h light:dark 18C, standing, 16h:8h light:dark 25C, shaking, continuous light Media Designed in this Study ASWH (Artificial Seawater for Halomonas) Per litre Sucrose NaCl Ultramarine synthetic sea salts Tricine Extra salts stock solution 10g 16.4g 33.6g (see below) 0.50g 3.75 ml (see below) Nutrient stock solution: Per litre NaNO3 Na2HPO4 K2HPO4 83.4g 1.20g 1.00g ‘Ultramarine synthetic’ sea salts Supplied by Waterlife Research Industries Ltd., 476 Bath Road, Longford West Drayton, Middlesex, UB7 0ED, England Euglena Minimal Medium Per litre Glutamic acid Glycine Glucose Malic acid MgSO4.7H2O CaCl2.2H2O CoCl2.6H2O Extra salts stock solution (from ASWH) Euglena salts Vitamin mix 3g 3g 3g 3g 500mg 200mg 25mg 5.00ml 0.5ml 1.5ml Adjust to pH 4.8 with 10M KOH Vitamin Mix: Per litre Biotin Thiamine HCl 40mg 40mg Euglena Salts: Per litre H3BO3 CuSO4.5H2O Fe(NH4)2SO4.6H2O MnSO4.4H2O ZnSO4.7H2O (NH4)6Mo7O24.4H2O 5.7mg 15.7mg 140mg 8.1mg 79mg 200mg Growth of algae at natural vitamin B12 concentrations To assess whether the concentration of vitamin B12 in the natural environment was limiting for the growth of vitamin B12-dependent freshwater and marine algae, Euglena gracilis, Amphidinium operculatum and Porphyridium purpureum were grown in media made exactly as described above, except that distilled water was replaced by natural filter-sterilised water. For the growth of freshwater algae, the water was collected from the Granta pond, Cambridge, UK and used to make the appropriate medium. The medium was then filter-sterilised through a 0.2 m Millipore filter before being used. Exactly the same protocol was used for the growth of marine algae, except the water used was seawater collected at West Runton, Norfolk, UK. Bacterial growth was not observed in the filter-sterilised media unless it was inoculated with a bacterial culture. Supplementation of Lobomonas rostrata medium Lobomonas rostrata was grown in 50 ml Jaworski’s medium for 7 days in continuous light with shaking in the presence of 10 g/l vitamin B12, 10 mM methionine and 1 M folic acid. A 200 l sample of this culture was removed and used to inoculate fresh medium, which was grown for a further 7 days. This subculturing was repeated 6 times. After each 7 day period, a 1 ml sample of culture was removed and the optical density measured at 600 nm. For homocysteine analysis, a 40 ml sample of culture, grown for 7 days, was centrifuged at 5,000 g for 20 min. The supernatant was discarded, and the pellet resuspended in 1 ml distilled water. The cells were sonicated with 6 x 20 second bursts separated by 30 s, on ice, in the presence of 100 l glass beads (213-300 m). The cell debris and glass beads were removed by centrifuging the sample at 13,000 g for 5 min at 4 C, and the supernatant was used for homocysteine analysis as described below. Homocysteine analysis Homocysteine levels were determined by HPLC after pre-column derivatisation with monobromobimane, as previously described2, with some modifications. Cell-free extracts were derivatised in a reaction mixture containing 0.1 M Hepes-NaOH, pH 8.0, 5 mM monobromobimane, 2 mM DTT (final concentrations). The reaction was incubated in the dark at room temperature for 10 min before the addition of 25 mM methanesulphonic acid. Samples were run on an Aglient 1100 series HPLC equipped with fluorescence detector (excitation 388 nm; emission 480 nm) using an Ace 5 AQ column (4.6 x 250 mm; Advanced Chromatography Technologies) at 37 ºC. A gradient of acetonitrile in 0.25 % acetic acid and 57 mM sodium perchlorate at a flow rate of 1 ml min-1 was used to elute the fractions. Thiols were identified by comparison against commercial standards. The concentration of homocysteine was standardised against total soluble protein in the extract. The concentration of soluble protein was determined using the Bradford assay. Isolation of bacteria from freshwater Samples of water were collected from the Granta pond, Cambridge, UK in sterile bottles. A 200 l sample of the water was spread onto LB agar plates, and the plates were incubated at 20 C for 48 h. Bacterial colonies, each with different morphologies, were picked from the plates with a sterile wire loop, re-cultured onto fresh LB agar plates, and incubated for a further 48 h. Individual colonies were then used to inoculate E. gracilis cultures. Porphyridium purpureum cell mass determination P. purpureum cells were grown in 50 ml PE medium for 28 days as described above. The entire culture was transferred to a 50 ml tube, and centrifuged at 5,000 g for 10 min. The pellet was resuspended in 1 ml fresh PE medium, and transferred to a pre-weighed 1.5 ml tube. The cells were centrifuged at 13,000 g for 5 min, and the supernatant discarded. The 1.5 ml tube, containing the cell pellet was re-weighed, and the cell mass determined. Identification of isolated bacteria Three colonies were picked from an agar plate and used to inoculate liquid media. The cells were grown to stationary phase. The culture was centrifuged at 4,500 g for 10 min, the pellet resuspended in 200 l sterile distilled water and heated to 100C for 15 min. The sample was cooled to room temperature, centrifuged at 13,000 g, and the supernatant was used as a crude cell extract for PCR. The 16S rRNA gene was amplified by PCR using degenerate primers as described previously3. The amplified product was gel purified, and directly sequenced using the same primers that were used for PCR amplification. The 16S rRNA gene sequence was then compared to other sequences in the ribosomal database at the Michigan State University (http://rdp.cme.sus.edu). DAPI staining A stock solution of 1 mg/ml DAPI (4’6-diamidine-2’-phenylindole dihydrochloride) was prepared in methanol. The stock solution was diluted to a final concentration of 1 g/ml with sterile seawater. An aliquot of 500 l of a Porphyridium purpureum culture was placed in an eppendorf tube, and the cells were left to settle for 30 min. The supernatant was removed, and 500 l of 1 g/ml DAPI was incubated with the cells for 15 min. The cells were washed twice with 1 ml sterile seawater, mounted on a glass slide and visualised under an eppifluorescent microscope at 325 nm, 400x magnification. The P. purpureum cells were not centrifuged at any point during the staining procedure as this disrupts the clumping of cells. Growth experiments with Halomonas sp. A single Halomonas sp. colony picked from an ASWH agar plate was used to inoculate 10 ml ASWH liquid medium, and the cells grown at 30 C for 16 h. A 2 ml sample of this culture was used to inoculate 100 ml ASWH medium containing 0.01% w/v fucoidin, and the cells were grown aerobically at 30 C. A 1 ml sample was taken every 4 h and the OD600 measured. After, 16, 24 and 36 hours a 10.1 ml sample was taken from the culture: 100 l was used for serial dilutions on ASWH agar plate, and the remaining 10 ml of medium was centrifuged at 4,000 g for 10 min at 20 C. The supernatant was discarded, the pellet resuspended in 500 l sterile water, and the resulting solution assayed for vitamin B12 (as above). Reverse Transcription – PCR Total RNA was extracted from C. reinhardtii or T. pseudonana using the method described4 and stored under ethanol at -80C. A 5-100 g sample of RNA was precipitated and treated with Rnase-free DNase (Promega) for 30 min at 37 C to remove any contaminating DNA. The cDNA was synthesized using Superscript IITM (Invitrogen) with dT17 primers exactly as described by the manufacturers instructions. The cDNA was amplified with Taq DNA polymerase (Bioline). The PCR conditions for the amplification of the metE fragment from C. reinhardtii were 28 cycles at 94 C for 40 seconds, 55 C for 1 minute, and 70 C for 1 minute. The metH and actin fragments from C. reinhardtii were amplified under exactly the same conditions except the annealing temperature was lowered from 55 C to 45 C. The primers used for PCR on C. reinhardtii cDNA were: metE F, 5`-GTGGACGACCCCCGCTCTGCG-3` metE R, 5`-CCATGCGGTCGATGGCGGGC-3` metH F, 5`-CAAGTACAAGGCGTGGAAGG-3` metH R, 5`-GTTGGTCTCAATGATGTCCG-3` actin F, 5`-TATCGTGCTGGACTCTGG-3` actin R, 5`-GATCTTCATCAGGTAATCG-3`. The PCR conditions for the amplification of the cbiP and actin fragments from T. pseudonana cDNA were 35 cycles at 94 C for 40 seconds, 48 C for 40 seconds, and 70 C for 1 minute. The primers used for the PCR on T. pseudonana cDNA were: TP cbiP F, 5`-TCTTGCTCAAGTCAGGAGGC-3` TP cbiP R, 5`-CACGGTACCACAAACACAGG-3` TP actin F, 5`-AACATTATTGTTATTGGGAATGAGC-3` TP actin R, 5`-ACTCTGGAAAGTAGAAAGAGAAGCC-3`. References 1. Troxler, R. F. Synthesis of bile pigments in plants. Formation of carbon monoxide and phycocyanobilin in wild-type and mutant strains of the alga, Cyanidium caldarium. Biochemistry, 11, 4235-4242 (1972) 2. Fahey, R. C. & Newton, G. L. Determination of low-molecular-weight thiols using monobromobimane fluorescent labeling and high-performance liquid chromatography. Methods in Enzymology 143, 85-96 (1987) 3. Lane, D. J. Nucleic Acid Techniques in Bacterial Systematics. E. Stackebrandt and M. Goodfellow, eds. N. Y., John Wiley and Sons: 115. (1991) 4. Witman, G. B., Carlson, K., Berliner, J. & Rosenbaum, J. L. Chlamydomonas flagella. I. Isolation and electrophoretic analysis of microtubules, matrix, membranes, and mastigonemes. J. Cell Biol. 54, 507-539 (1972)