Supporting Information
advertisement

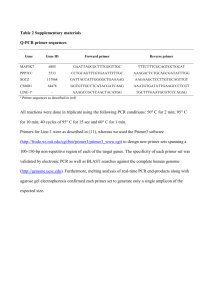
Supporting Information Experimental Procedures Bacterial strains and culture conditions Bacterial strains and plasmids used in this study are listed in Table 3. Bacteria were grown in Lysogeny Broth (LB) broth or on LB agar (Sambrook et al., 1989) at 37°C for E. coli and at 25°C for Burkholderia sp. Ha185. The 16S rRNA gene amplification was carried out using primers U16a and 1087R (Table S1) as described in Wang and Wang (1996). Amplification of products of the correct size (~ 1,600 bp) was confirmed by sequencing on an Applied Biosystems 3730xl and 9 ABI 3700 DNA Analyzer by Macrogen sequencing services (http://dna.macrogen.com/eng/). Cultures were incubated with shaking at 250 rpm. Antibiotic concentrations (μg ml-1) used for Burkholderia sp. Ha185 were as follows: ampicillin 100, gentamicin 20, kanamycin 50, and tetracycline 30. Concentrations used for E. coli were: ampicillin 100, gentamicin 20, kanamycin 50, and tetracycline 15 (Table 3). Draft Burkholderia sp. Ha185 genome sequence, annotation, and sequence alignment Genomic DNA (gDNA) was extracted using a Genomic DNA prep kit (SGD41-C100, SolGent, Daejeon, Korea) in accordance with the manufacturer’s instructions. A low-coverage draft Burkholderia sp. Ha185 genome was sequenced and assembled by Macrogen (Seoul, South Korea) using a GS-FLX Titanium next generation sequencing platform (454 Life Sciences, Branford, CT, USA). Sequencing generated 199,790 reads (8,549,297 bp) that were de novo assembled into 1,718 contigs with an average size of 4,976 bp. To identify proteins of interest from the Burkholderia sp. Ha185 genome, a BLAST database containing the contig files of the draft genome sequence was constructed using the makeblastdb tool of the blast+ program (Version 2.2.28) (Altschul et al., 1990). The nucleotide database was then translated into all six possible reading frames using tBLASTn software (blast+ version 2.2.28). The query protein sequences were then downloaded from UniProt (Apweiler et al., 2013) (http://www.uniprot.org). The tBLASTn program was used to search the Burkholderia sp. Ha185 genome protein database using a protein query in either the predicted ORFs or the nucleotide sequence database. The nucleotide sequence surrounding the region of a hit was extracted from the database using the extractseq suite from EMBOSS (version 6.5.0) (Rice et al., 2000). Sequence alignments were carried out using Geneious version 6.1.5 (Biomatters, Auckland, New Zealand). Predicted ORFs were annotated using BLASTx and BLASTp searches in GenBank (Table 2). Amino acid sequences were also compared against the Protein Data Bank, InterPro, Pfam, and SwissProt databases using the MyHit Motif Scan and TMpred search tools (http://myhits.isb-sib.ch/cgi-bin/motif_scan). Construction of a mini-Tn5Km1 transposon mutant library and mutant selection Transposon mutagenesis was performed using the mini-Tn5 derivative Tn5Km1, as described by de Lorenzo et al. (1990). To allow antibiotic selection of the recipient strain, the plasmid pBBR1MCS-5 was transformed into Burkholderia sp. Ha185. The mini-Tn5 Km1 mutants were screened for loss of phosphate solubilization by measuring reduced zones of clearance after incubation at 25°C for 14 days on HA agar plates (55.5 mM glucose, 62.5 mM NH4NO3, 67.1 mM KCl, 2.0 mM MgSO4·7H2O, 6.6 µM MnSO4·H2O, 6.6 µM FeSO4·H2O, 6.0 g Ca10(PO4)6(OH)2, and 1.5% (w/v) agarose agar; pH 6.5. One-hundred microliters of 10× trace element solution were added after autoclaving. Trans-complementation of the hemX::Tn5 mutant The primer pair HemCF/HemXR (Table S1) was designed to amplify a region beginning 299 bp 5′ of hemC, encompassing the putative promoter region, and ending 409 bp 3′ of hemX (Fig. 1A). For amplification of the hemX gene, HemXF was used in place of HemCF (Fig. 1A). The resultant amplicons were cloned into pGEM-T Easy (Promega BioSciences, San Luis Obispo, CA, USA), and cloned inserts were end-sequenced using the M13F and M13R primers. Each of the validated cloned inserts were cleaved from pGEM-T Easy using BamHI, and independently ligated into the compatible BglII site of pME6010, from where they were then independently transformed into E. coli DH10B. The validated clones, designated pHEMCX (hemC-hemX) and pHEMX (hemX), were separately electroporated into hemX::Tn5. The trans-complemented hemX::Tn5 derivatives were then assessed for restoration of the phosphate-solubilizing phenotype on HA agar plates by spotting 1 µl of bacterial culture (containing approximately 105 colony forming units (CFU) onto a HA agar plate. Plates were incubated for 14 days at 25°C to allow the development of zones of clearance (Fig. 1A). Wild-type Burkholderia sp. Ha185 and the hemX::Tn5 mutant were used as positive and negative controls, respectively. Hydroxyapatite solubilization by Burkholderia sp. Ha185 and derivative mutants grown in HSUHA liquid culture. For liquid culture assay, Burkholderia sp. Ha185 and its derivatives were grown in LB broth for 48 h at 25°C with appropriate antibiotics until the OD600 reached approximately 4.0. Each isolate was then pelleted by centrifugation for 3 min at 5900 × g at 22°C. The cell pellets were resuspended in 100 µl of HSU liquid medium (55.5 mM glucose, 1.38 mM K2SO4, 2.63 mM MgCl2, 4.67 mM NH4Cl, 0.27 mM CaCl2, 25.0 mM NaCl) and independently used to inoculate 50- ml HSU broth cultures supplemented with 0.3 g of HA, resulting in an approximate cell concentration of 1.0 × 106 CFU/ml. The HSU-HA cultures were then incubated for 72 h. At 24, 48, and 72 h post-inoculation, CFU were determined by serial dilution. For the assessment of pH, soluble P, and organic anion content, 1 ml of cell culture was pelleted by centrifugation at 15,700 × g for 10 min. The supernatant was filtered using a 0.22-µm polyvinylidene fluoride (PVDF) syringe filter (13 mm diameter, Thermo Fisher Scientific, Waltham, MA, USA). The pH of the bacterial filtrate was determined using a Handylab pH meter (SCHOTT Instruments, Waltham, MA) at 22°C. Free phosphate was quantified using the method of Murphy and Riley (1962). To determine whether cobalamine is directly involved in P solubilization, cobalamine sourced from Sigma (CAS 68-19-9) was exogenously supplied to a HSU liquid culture of the hemX::Tn5 mutant or wild-type Burkholderia sp. Ha185 at a final concentration of 1.4 nM. Assessment of extracellular and intracellular soluble phosphate and organic anion profiles At 72-h post-inoculation in liquid culture, the amount of intracellular soluble phosphate (PO43−), was determined by assessing the sonicated filtrate derived from 1 ml of cells. Cells were centrifuged for 1 min at 15,700 × g, the supernatant was discarded, and the pellet was resuspended in 1 ml of HSU medium without HA and glucose. The resuspended cell pellets were independently sonicated three times for 30 s on wet ice, with 30-s intervals between pulses, using a Soniprep 150 Ultrasonic Disintegrator (Sanyo Gallenkamp PLC, Leicester, UK). The sonicated samples were centrifuged at 15,700 × g for 10 min to pellet the cell lysate, and the supernatants were filtered using a 0.22-µm PVDF syringe filter. Filtrates were stored at −20°C. The concentration of soluble phosphate in sonicated filtrates was assessed, and the organic anion profiles determined by HPLC as outlined below. Further liquid culture assays were performed substituting glucose with 54.9 mM mannitol as the sole carbon source. Soluble phosphate (90.12 mM Na2HPO4 and 22.04 mM KH2PO4 in 50 ml) was also used in place of HA as the only source of phosphate as the negative control. All experiments were performed three times independently. An uninoculated HSU medium-only negative control was included in all assays. Organic anion profiling by HPLC Organic anion profiles of Burkholderia sp. Ha185 and its variant mutants were analyzed and quantified by HPLC as described previously (Giles et al., 2014) with modifications as described below. HPLC was conducted using a Shimadzu LC system (Shimadzu Corporation, Kyoto, Japan). Standard stock solutions of D-gluconate sodium salt (GA, 99%), 2-keto-D-gluconate hemicalcium salt hydrate (2-KGA, 100%), 5-keto-D-gluconate potassium salt (5-KGA, 98.0%), pyruvate (PA, 98%), malonate (MA, 99%), shikimate (SA, 99%), L-malate (MA, >99%), DLlactate (LA, 85%), formate (FA, 98%), and acetate (100%) were prepared by dissolving in HSU liquid culture medium. A Rezex ROA-Organic Acid H+ (8%) column (3000 × 7.8 mm, Phenomenex, Torrance, CA, USA) with a guard column (Carbo-H 4 × 3.0; Phenomenex) was used to separate and quantify organic anions. The column contained sulfonated styrenedivinylbenzene spheres in 8% cross-link resin with hydrogen ionic form. Sulphuric acid (5 mM) was used as the mobile phase and was prepared by filtering through a 0.45-µm cellulose acetate membrane (47 mm; Advantec MFS Inc, Dublin, CA, USA). Each sample was filtered through a 0.22-µm PVDF syringe, and was either stored undiluted or diluted 10- or 20-fold in HSU medium at 4°C prior to analysis. Samples were injected (20 µl) at a flow rate of 0.5 ml/min, with a column temperature of 55°C and a detection wavelength of 210 nm. Organic anions were detected by comparing the retention time of sample to standards. Samples were quantified by the peak height of chromatograms using an external calibration standard curve. All data were processed using LC solution software. Identification of transposon insertion sites by touchdown PCR To identify the mini-Tn5Km1 transposon insertion sites within the genomes of the mutants, genomic DNA (gDNA) was isolated from each mutant using a Genomic DNA Prep kit (SGD41C100, SolGent, Daejeon, Korea) following the manufacturer’s instructions. Genome walking was then performed using a modified method from Guo and Xiong, (2006). The transposon insertion site was identified by sequencing the product of a restriction site PCR using a touchdown PCR protocol. The mini-Tn5Km1 transposon-specific primers are listed in Table S1. The PCR amplification was carried out using Platinum Taq DNA Polymerase High Fidelity (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. The first round of PCR was performed using 0.2 µM of the Sp1 primer and each one of the universal primers individually, in different reactions. Each reaction contained 0.2 µM gDNA, 10× High Fidelity reaction buffer, 0.2 mM dNTPs, 2 mM MgCl2, 1 U Platinum Taq DNA polymerase, and distilled H2O in a total volume of 20 µl. Touchdown PCR parameters were 94°C for 2 min; three cycles of 94°C for 20 s, 60°C for 20 s, and 68°C for 2 min; three cycles of 94°C for 20 s, 57°C, 55°C, and 45°C for 20 s, and 68°C for 2 min; 35 cycles of 94°C for 20 s, 50°C for 20 s, and 68°C for 2 min; 68°C for 5 min. Reactions were then held at 4°C prior to the next round of PCR. The products obtained from the first round of PCR were used as templates for the second round of touchdown PCR. Either Sp2A or Sp2B was used as the forward primer, and T7 was used as the reverse primer. The T7 universal primer binds to the T7 tail sequence of the RSO primer incorporated during the first run. The second round of PCR was carried out as follows: 94°C for 20 s, 60°C for 20 s, 68°C for 2 min; three cycles of each temperature at 57°C and 55°C, then 68°C for 2 min; 35 cycles of 94°C for 20 s, 50°C for 20 s, and 68°C for 2 min; 68°C for 5 min. The PCR products were electrophoresed on a 1% agarose gel, and samples with bands >300 bp were then purified using a High Pure PCR Purification kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. Purified PCR products were cloned into pGEM®-T Easy using the LigaFastTM Rapid DNA Ligation System (Promega, Madison, WI, USA) according to manufacturer’s instructions. The resulting plasmids were validated by restriction enzyme profiling using EcoRI. Plasmid DNA was isolated using a High Pure plasmid isolation kit (Roche) in accordance with the manufacturer’s instructions, and then sequenced using M13F and M13R primers. Primer sequences are listed in Table S4. Sequencing was performed by Macrogen (Seoul, Korea). At least two cloned PCR amplicons were sequenced for each product. Each sequence was aligned to confirm the presence of mini-Tn5Km1 using Sequencher® version 5.1 (Gene Codes Corporation, Ann Arbor, MI, USA). Databases at the National Center for Biotechnology Information were searched using BLASTx in the reference proteins database (Altschul et al., 1997; Schäffer et al., 2001). Isolation of RNA from Burkholderia sp. Ha185 and synthesis of complementary DNA To quantify the expression of selected genes throughout the growth cycle of Burkholderia sp. Ha185 in HSU-HA medium, wild-type Burkholderia sp. Ha185 was incubated in liquid medium containing glucose. Duplicate cultures were grown in HSU-HA medium with mannitol as the sole carbon source (HSU-HA-mannitol), and in HSU with soluble phosphate instead of HA (HSU-phosphate). Total cellular RNA was isolated from cultures using RNAprotect Bacteria Reagent and an RNeasy Mini Kit (Qiagen, Hilden, Germany) at 24, 36, 40, 48, and 72 h postinoculation. Samples from each cell culture (200 µl) were independently aliquoted into 1.5-ml tubes containing 400 µl of RNAprotect Bacteria Reagent. The samples were then processed following the protocol supplied by the manufacturer. Pellets were stored at −80°C or used for immediate RNA extraction following the RNeasy Mini Kit protocol. On-column DNase digestion was performed using 3 µl of recombinant RNase-free DNase I (Roche) with 77 µl of 10× incubation buffer per column, and incubated at room temperature for 15 min. RNA was then eluted in RNase-free water using the RNeasy Mini Kit protocol. The concentration of the resultant RNA was measured using a Nanodrop ND-1000 spectrophotometer (Eppendorf, Hamburg, Germany), and the purity and integrity were confirmed by 1% agarose gel electrophoresis. Purified RNA was again treated with RNase-free DNase and used as a template for cDNA synthesis using reverse transcriptase. RNase-free DNase (Roche, 0.5 µl) was added to 0.8 µl DNase buffer and 6.7 µl RNA. Samples were gently mixed using a pipette and incubated at 37°C for 15 min. Samples were centrifuged briefly and then RNase-free EDTA was added to a final concentration of 8 mM (pH 8.0) (Life Technologies), followed by incubation at 75°C for 10 min. Following incubation, samples were centrifuged briefly and then placed on ice prior to cDNA synthesis. The concentration of DNase-treated RNA was measured again by Nanodrop spectrophotometer, and the purity and integrity were confirmed by 1% agarose gel electrophoresis. Reverse transcription PCR reactions were performed using a Transcriptor High Fidelity cDNA Synthesis Kit (Roche) according to the manufacturer’s instructions. A final volume of 11.4 µl, containing 60 µM random hexamer primers, ~100 ng DNase-treated RNA, and 7.4 µl of PCR-grade water, was incubated at 65°C for 10 min. Reactions were then centrifuged briefly prior to cDNA synthesis. The following were then added to each reaction: 1× Transcriptor High Fidelity Reverse Transcriptase Reaction Buffer, 5 mM DTT, 20 U Protector RNase Inhibitor, and 10 U Transcriptor High Fidelity Reverse Transcriptase, to give a final volume of 20 µl. Reactions were gently mixed by pipetting and then incubated at 53°C for 30 min in a Mastercycler EP thermal cycler, followed by 85°C for 5 min to inactivate the transcriptase. The cDNA was stored at −80°C. Quantitative reverse transcriptase PCR from cDNA template To define the relative RNA levels of hemX and its neighboring genes, including hemC and ppc, in Burkholderia sp. Ha185, qRT-PCR was carried out using the reference genes gltB, recA, and gyrB. At various time points, expression of these genes in cells grown in HSU-HA liquid culture medium was compared with expression in HSU-phosphate culture medium. The qRT-PCR reactions were performed using a Corbett Rotor-Gene 6000 cycler (Qiagen). All reactions contained 1× SensiMix SYBR No-ROX mix (BioLine, London, UK), approximately 50 ng of template DNA (equivalent RNA concentration as measured by Nanodrop ND-1000 spectrophotometer), 250 nM of each primer, and DNase/RNase-free water, in a final volume of 10 µl. Cycling parameters for qRT-PCR were 50°C for 2 min; 95°C for 10 min; 35 cycles of 95°C for 15 s and 60°C for 15 s; 72°C for 15 s. Data was acquired at the end of each cycle. A high resolution melting curve (temperature gradient of 50–95°C) was generated postamplification to identify amplified products, and to distinguish PCR amplicons from primer dimers or background artefacts. Primers used in this study are listed in Table S3. Raw qRT-PCR data were imported directly from the thermal cycler, and the R-value and PCR efficiency of each primer set were determined using LinRegPCR (Ramakers et al., 2003). Raw data were also analyzed using REST-MCS version 2 software (Pfaffl, 2001), allowing comparison of the target gene, bxpC, with three reference genes, gltB, recA, and gyrB. Statistical analysis All numerical data are expressed as the mean of three replicates ± the standard error of the mean (mean ± SEM). One-way analysis of variance (ANOVA) was used to calculate significant differences between treatments (*P < 0.05, ** P < 0.01, *** P < 0.001). Generalized Linear Modelling was used to generate pair-wise correlations between soluble phosphate released over time, culture filtrate pH, and soluble phosphate and 2-KGA concentrations in the cytosol. Differences between treatment groups were tested using the Tukey method at the 5% level. Analyses were performed using Minitab version 15 (Minitab, Inc., www.minitab.com). Results Fig. S1. Schematic diagram of the proposed haem biosynthesis pathway. hemX codes for a bifunctional uroporphyrinogen-III synthetase/uroporphyrin-III C-methyltransferase, which is hypothesized to synthesize precorrin-1 (dashed arrow) via the intermediate uroporphyrinogen III (green dashed bracket). Precorrin-2 (highlighted in red box) is an important branch point, leading to biosynthesis of different tetrapyrrole compounds, cobalamine (vitamin B12), coenzyme F430, siroheme, heme, and heme d1. Fig. S2. Co-occurrence analysis of Burkholderia spp. PPC, HemC, HemX, and HemY regions across different bacterial families using STRING 9.05. B. pseudomallei K96243 and B. multivorans 17616 (highlighted in purple box) were used as the consensus amino acid sequences. The occurrence pattern is colored from white to black, where no homology is indicated by white squares and 100% homology is indicated by black squares. Fig. S3. ClustalW alignment of FAD-dependent gluconate 2-dehydrogenase (subunit I) from Erwinia cypripedii ATCC 29267, Burkholderia phytofirmans PsJN, and protein sequences from Burkholderia sp. Ha185 (Contig00115). The FAD binding site is highlighted in a purple box (consensus amino acid residues 9–42, [VD]-X-[V]-X(2)-[G]-X[GW]-X-[G]-X-[I]-X(3)-[EL]-X(3)-[GL]-X-[VV]-X-[LERG]-X(2)). The red box indicates a glucose-methanol-choline oxidoreductase domain, N-terminal (GMC_OxRdtase_N) domain, and the glucose-methanol-choline oxidoreductase. The C-terminal (GMC_OxRdtase_C) domain is highlighted in the green box. Sequences from B. phytofirmans PsJN (rows 1–3) are identified by the protein accession number (GenBank database), followed by the protein entry name (UniProt database). Sequences derived from the Burkholderia sp. Ha185 draft genome (row 4) are identified by the contig number. Protein alignment was carried out using the BLOSUM-62 matrix, where black highlighting indicates 100% similarity, dark grey is 80–100% similarity, light grey is 60–80% similarity, and white is < 60% similarity. Fig. S4. ClustalW alignment of heme-dependent gluconate 2-dehydrogenase (acceptor) (subunit II) from Burkolderia phytofirmans strain PsJN and protein sequences from Burkholderia sp. Ha185. The predicted heme-binding motif is highlighted in the purple box (consensus amino acid residues 96–100, 266–270, and 415–419), where the CXXCH amino acid motif is highly conserved in the center of a cytochrome c domain. Three cytochrome c units are found in gluconate 2-dehydrogenase (acceptor) (HEM-GADH), and are highlighted in red boxes. Sequences from B. phytofirmans PsJN (rows 1–10) are identified by the protein accession number (GenBank database), followed by the protein entry name (UniProt database). Sequences derived from the Burkholderia sp. Ha185 draft genome (rows 11–17) are identified by the contig numbers. Fig. S5. ClustalW alignment of gluconate 2-dehydrogenase (acceptor) (subunit III) from Burkholderia phytofirmans strain PsJN, Erwinia cypripedii, and protein sequences from Burkholderia sp. Ha185. The twin arginine translocation (Tat) signal profile (Prosite PS51318) is located at residues 1–50 in the consensus sequence. The signal peptide motif is highlighted in a green box, with the consensus sequence [ST]-R-R-X-F-L-X and a short AXA motif of each protein sequence underlined in red. Sequence from E. cypripedii (row 1) is identified by the protein accession number (GenBank database). Sequences from B. phytofirmans PsJN (rows 2–4) are identified by the protein accession number (GenBank database), followed by the protein entry name (UniProt database). The sequence derived from the Burkholderia sp. Ha185 draft genome (row 5) is identified by the contig number. Protein alignment was carried out using the BLOSUM-62 matrix, where black highlighting indicates 100% similarity, dark grey is 80–100% similarity, light grey is 60–80% similarity, and white is < 60% similarity. Fig. S6. Burkholderia sp. Ha185 Contig00447 and Contig00115 (dark purple) are homologous to the Burkholderia phytofirmans PsJN gadh operon (chromosome 1; 241,745–246,148 bp, light purple). gadh(I), gadh(II), and gadh(III) code for FAD-dependent GADH, heme-dependent GADH, and GADH protein, respectively. Fig. S7. ClustalW alignment of partial heme-dependent gluconate 2-dehydrogenase (acceptor) sequences from B. phytofirmans strain PsJN and Burkholderia sp. Ha185 (81–160 aa residues). The predicted heme-binding motif CXXCH is highlighted in a black box (consensus amino acid residues 96–100), and shows the motif is highly conserved. Sequences from B. phytofirmans PsJN (rows 1–10) are identified by the protein accession number (GenBank database), followed by the protein entry name (UniProt database). Translated sequences derived from the Burkholderia sp. Ha185 draft genome are identified by the contig numbers (rows 11–16). Protein alignment was carried out using the BLOSUM-62 matrix, where black highlighting indicates 100% similarity, dark grey is 80–100% similarity, light grey is 60–80% similarity, and white is < 60% similarity. Table S1. Oligonucleotide primers used in this study. Purpose Name Sequence (5′ - 3′)* Gene complementation HemCF HemXR HemXF HemXR2 AAAGGATCCGACTGCAA TGTGAACAGCAC AAAGGATCCTATTCGT GCATGCGATGCGC AAAGGATCCGCTGCA TCACGAACACAC AAAGGATCCTACAGCAGGATGAACAGCACCA CC ATCGACGAAGCCGACTGGCTCGATGC TCTCGCGATAATGCTGGCTCGCC Primer walking 342F 042R qRT-PCR primer sets qGyrBF qGyrBR qRecAF qRecAR qGltBF qGltBR qHemXF qHemXR qPpcF qPpcR qHemCF qHemCR GGATGAGCGGTATTTGAAGG ATATGCACGAGCCAACTCAC CGTATCGGTTCGATCAAGAAG CGAAATACCTTCGCCATACAG GATCTTCGCGATGTCCTTG GGACACGAACAACATCAACC CACGCATGACTGAAACGAC ATCACGACGACAAACCACAG CATTCACGAAGCGGTGATCG GGAATACTCGGCATACGGCA ATCCTCGGAATGACGACACG ACGTCTTTGAGCGAATGCAC Gene sequencing M13F† M13R† GTAAAACGACGGCCAGT GCGGATAACAATTTCACACAGG *Underscore denotes designed restriction site †Oligonucleotide from Macrogen universal primer list References Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J Mol Biol 215: 403-410. Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389−3402. Apweiler, R., Martin, M.J., O'Donovan, C., Magrane, M., Alam-Faruque, Y., Alpi, E., Antunes, R., Arganiska, J., Casanova, E.B. and Bely, B. (2013) Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res 41: D43-D47. Consortium, U. (2014) Activities at the universal protein resource (UniProt). Nucleic Acids Res 42: D191−D198. Giles, C.D., Hsu, P.-C., Richardson, A.E., Hurst, M.R.H., and Hill, J.E. (2014) Plant assimilation of phosphorus from an insoluble organic form is improved by addition of an organic anion producing Pseudomonas sp. Soil Bio Biochem 68: 263-269. Guo, H. and Xiong, J. (2006) A specific and versatile genome walking technique. Gene 381: 18-23. de Lorenzo, V., Herrero, M., Jakubzik, U., and Timmis, K.N. (1990) Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J Bacteriol 172: 6568-6572. Murphy, J. and Riley, J. (1962) A modified single solution method for the determination of phosphate in natural waters. Anal Chim Acta 27: 31-36. Pfaffl, M.W. (2004) Quantification strategies in real-time PCR, p 87-112. In Bustin SA (ed), Quantification strategies in real-time PCR, AZ of quantitative PCR, Chap 3, 87−112. La Jolla, CA. Ramakers, C., Ruijter, J.M., Deprez, R.H.L., Moorman, A.F. (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339: 62−66. Rice, P., Longden, I., Bleasby, A. (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16: 276−277. Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989) Molecular cloning. Vol. 2. New York, USA. Cold spring harbor laboratory press. Schäffer, A.A., Aravind, L., Madden, T.L., Shavirin, S., Spouge, J.L., Wolf, Y.I., Koonin, E.V., Altschul, S.F. (2001) Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 29: 2994−3005. Wang, G.C. and Wang, Y. (1996) The frequency of chimeric molecules as a consequence of PCR co-amplification of 16S rRNA genes from different bacterial species. Microbiology 142: 1107-1114.