WORD document
advertisement

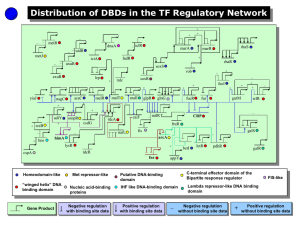
S.2: Supplementary Material – Description of Algorithms The gene regulatory network is represented as a directed graph G=(V, E) where each protein in the network is a vertex (v V). The protein can be a transcription factor (tf) or a regulated gene (rg) where tf, rg V. The transcriptional interaction (from a transcription factor to the regulated gene) is a directed edge (e E). The network is implemented using two hash tables PR (Parent of a Regulated gene) and CT (Child of a Transcription factor). PR consists of |V| keys, one for each vertex in V. The key is a regulated gene and the values correspond to all the transcription factors regulating it (i.e. for each key rg, the value corresponds to all vertices tf such that there is an edge (tf → rg) E). CT consists of |V| keys, one for each vertex in V. The key is a transcription factor and the values correspond to all genes that it regulates (i.e. for each key tf, the value corresponds to all vertices rg such that there is an edge (tf → rg) E). Each protein (v V) is characterised by a domain architecture, i.e. the ordered set of domains from the N-terminus to the C-terminus. Proteins with the same domain architecture, ignoring gaps and internal duplications, are considered homologues in this analysis. This information is stored in a hash table DA of |D| elements, one for each domain architecture d. A key corresponds to a domain architecture d and values correspond to all vertices h (homologous proteins) in the graph that has the domain architecture d. In other words, this hash table represents genes (h) that have arisen by duplication from a common ancestor. Another hash table DAG consists of |V| keys, one for each vertex in V such that the key is a gene and the value is its domain architecture, d. An explanation for each line in the algorithm for section S.2.1 is given as a guide. S.2.1: Characterization of the vertices in the network A. Algorithm to identify duplicated genes (regulated genes) that are controlled by the same transcription factors 01: for each d DA 02: for each h DA [d] 03: for each tf PR [h] 04: X [tf] ← 0 05: for each h DA [d] 06: for each tf PR [h] 07: X [tf] ← X [tf] + 1 08: for each h DA [d] 09: for each tf PR [h] 10: if X [tf] > 1 11: print (h) # for each domain architecture (d) in DA # for each homologous gene (h) with domain architecture (d) # for each tf that regulates h # set the count X [tf] to 0 # for each homologous gene (h) with domain architecture (d) # for each tf that regulates h # increment the count X [tf] # for each homologous gene (h) with domain architecture (d) # for each tf that regulates h # if the tf regulates more than one homologous gene # print the rg B. Algorithm to identify duplicated genes (transcription factors) that control the same target genes 01: for each d DA 02: for each h DA [d] 03: for each rg CT [h] 04: X [rg] ← 0 # for each domain architecture (d) in DA # for each homologous gene (h) with domain architecture (d) # for each rg that is regulated by h # set the count X [rg] to 0 05: 06: 07: 08: 09: 10: 11: for each h DA [d] for each rg CT [h] X [rg] ← X [rg] + 1 for each h DA [d] for each rg CT [h] if X [rg] > 1 print (h) # for each homologous gene (h) with domain architecture (d) # for each rg that is regulated by h # increment the count X [rg] # for each homologous gene (h) with domain architecture (d) # for each rg that is regulated by h # if the rg is regulated by more than one homologous tf # print the tf S.2.2: Characterization of the edges in the network A. Algorithm to identify edges that have evolved by the duplication of RGs: 01: for each d DA 02: for each h DA [d] 03: for each tf PR [h] 04: X [tf] ← 0 05: for each h DA [d] 06: for each tf PR [h] 07: X [tf] ← X [tf] + 1 08: for each h DA [d] 09: for each tf PR [h] 10: if X [tf] > 1 11: print E (tf → h) B. Algorithm to identify edges that have evolved by the duplication of TFs: 01: for each d DA 02: for each h DA [d] 03: for each rg CT [h] 04: X [rg] ← 0 05: for each h DA [d] 06: for each rg CT [h] 07: X [rg] ← X [rg] + 1 08: for each h DA [d] 09: for each rg CT [h] 10: if X [rg] > 1 11: print E (h → rg) As mentioned in the manuscript, Algorithms A and B have precedence over C, meaning cases picked up by A and B are excluded from consideration by C. C. Algorithm to identify edges that have evolved by duplication of TFs and RGs: 01: for each d DA 02: for each h DA [d] 03: X [d] ← X [d] + 1 04: for each rg CT [h] 05: Y [ DAG [rg] ] ← Y [ DAG[rg] ] + 1 06: Z [rg] ← Z [rg] + 1 07: if X [d] > 1 08: for each rg CT [h] 09: if Z [rg] = 1 and Y [ DAG[rg] ] > 1 10: print E (h → rg) As described in the manuscript, Algorithms A, B and C have precedence over D, E meaning cases picked up by A, B and C are excluded from consideration by D and E. D. Algorithm to identify edges that have evolved by duplication of TFs/RGs but lost their old interactions and gained new interactions: D.1 Edges where the RG duplicates and gets regulated by a new TF and loses old interactions: 01: for each d DA 02: for each h DA [d] 03: X [d] ← X [d] + 1 04: for each tf PR [h] 05: Y [tf] ← 0 06: for each h DA [d] 07: for each tf PR [h] 08: Y [tf] ← Y [tf] + 1 09: for each h DA [d] 10: for each tf PR [h] 11: if Y [tf] = 1 and X [ DAG[h] ] > 1 12: print E (tf → h) D.2 Edges where the TF duplicates and regulates new RGs and loses old ones: 01: for each d DA 02: for each h DA [d] 03 X [d] ← X [d] + 1 04: for each rg CT [h] 05: Y [rg] ← 0 06: for each h DA [d] 07: for each rg CT [h] 08: Y [rg] ← Y [rg] + 1 09: for each h DA [d] 10: for each rg CT [h] 11: if Y [rg] = 1 and X [ DAG[h] ] > 1 12: print E (h → rg) E. Algorithm to identify edges that have evolved by innovation of new regulatory interactions: 01: for each d DA 02: for each h DA [d] 03: X [d] ← X [d] + 1 04: for each d DA 05: for each h DA [d] 06: if X [d] = 1 07: for each rg CT [h] 08: if X [ DAG [rg] ] = 1 09: print E (h → rg) S.2.3: Identification of network motifs The algorithms to identify Feed-forward motifs and Single Input Modules are described below. In these algorithms, P is an array which contains a list of transcription factors that regulate genes that are regulated by only one transcription factor and u, v, w V A. Feed Forward motif 01: for each u CT 02: X [u] ← 0 03: for each v CT [u] 04: X [v] ← 1 05: for each w CT [v] 06: X [w] ← X [w] + 1 07: if X [w] = 2 & (u ≠ v, u ≠ w, v ≠ w) 08: print E (u → v → w) 09: X [w] ← 1 B. Single Input Module motif 01: for each rg PR 02: for each tf PR [rg] 03: X [rg] ← X [rg] + 1 04: for each rg PR 05: if X [rg] = 1 06: Push (P, PR [rg]) 07: for each tf P 08: for each rg CT [tf] 09: if X [rg] = 1 10: print E (tf → rg) S.2.4: Identification of directly and indirectly regulated genes The breadth-first search1 method, which uses a recursive algorithm, was implemented to identify all nodes that can be reached from a given node. This was used to identify all other genes that can be affected by a transcription factor. The hash table CT provides a list of all directly regulated genes. The difference between the two sets gives the indirectly regulated genes by a transcription factor. References: 1. Introduction to Algorithms, 2nd Edition (2001), Cormen, T.H., Leiserson, E.E., Rivest, R. L. and Stein, C. The MIT Press. S.2.5: Simulation procedure to estimate the significance of duplication in the network The domain architecture for each transcription factor is stored in a hash table DATF of |D| elements, one for each domain architecture d. A key corresponds to a domain architecture d and values correspond to all vertices w that are transcription factors and have the domain architecture d. In other words, this hash table represents groups of homologous transcription factors. Similarly a hash table DARG is created where a key corresponds to a domain architecture and the values correspond to all vertices w that are target genes and have the domain architecture d. is a function that assigns a random domain architecture to a gene. However it is ensured that the number of genes with each domain architecture is the same as seen in the real network (as implemented in the hash tables). Randomise A. Calculation of P-value for the duplication of target genes (RGs) that are controlled by the same transcription factor (TF) 01: for i = 1 to 10,000 02: Randomise (DARG) 03: Perform S.2.1.A and count the number of RGs that satisfy the criteria 04: if the number of RGs in simulation ≥ number seen in the real network 05: n ← n+1 06: P-value = n/10,000 B. Calculation of P-value for the duplication of transcription factors that control the same target genes 01: for i = 1 to 10,000 02: Randomise (DATF) 03: Perform S.2.1.B and count the number of TFs that satisfy the criteria 04: if the number of TFs in simulation ≥ number seen in the real network 05: n ← n+1 06: P-value = n/10,000 S.2.6: Simulation procedure to estimate the significance of duplication in determining network topology 01: for i = 1 to 10,000 02: Randomise (DARG) 03: for each TF in CT 04: if number of distinct domain architectures of it’s RG ≤ number seen in the real network 05: n ← n+1 06: P-value = n/10,000