法布瑞氏症以高血壓和蛋白尿表現: 案例報告及文獻回顧 Fabry
advertisement

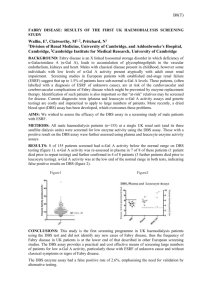
法布瑞氏症以高血壓和蛋白尿表現: 案例報告及文獻回顧 Fabry disease presented with hypertension and proteinuria:A Case Report and Literature Review 陳河卿 蔡宗諭 林如立 黃文宏 賴彬卿 Ho-Ching Chen, Tsung-Yu Tsai, Ru-Li Lin, Wen-Hong Huang, Bin-Ching Lai 長庚紀念醫院林口分院 Chang Gung Memorial Hospital, Linkou department, Taoyun, Taiwan Fabry disease is an X-linked inborn disorder caused by lysosomal-galactosidase A deficiency which leads to incomplete metabolism and lysosomal accumulation of glycosphingolipids, mainly globotriaosylceramide (GL3) in the lysosomes of various cell types and tissues and progressive kidney, cardiac, and neurologic involvement that can cause death in the fifth decade. A 32-year-old man visisted CV OPD at our hospital on March 06, 2009 due to hypertension (Blood pressure: 183/92 mmHg) noted since he was in his twenties. No treatment had been given before. Exams revealed regular heart beat but with sustained point if maximal impulse and EKG showed left ventricular hypertrophy. Cardiac echo revealed non-obstructive type hypertrophic cardiomyopathy. He was referred to a nephrology department with discovered proteinuria (0.8g per day) and hyperlipidemia (Cholesterol 224mg/dL). General findings were unremarkable : his weight was 60 kg, serum creatinine was 0.94 mg/dL, RPR/VDRL, ANA, HBV, HCV were negative, IgA was 201mg/dL and ASLO <52.80 IU/mL all in normal limit. He received renal biopsy on March 2, 2010. Norteworthly, his mother and his two uncle were all died due to stoke and his father also has hypertension. Kidney pathology in light microscopy revealed focal segmental glomerulosclerosis and many glomeruli have foam cells. Electron microscopy showed vacuolated podocytes contained abundant lipid inclusions which stain positive with colloidal iron and methylene. These inclusions composed of concentric layers with an onion skin appeareance which are considered a hallmark of glycolipid storage disorder. Fabry disease is diagnosed and proofed by low alpha-Gal A activity and gene mutation analysis. The Fabry nephropathy is characterized by initial proteinuria in the second to third decades of life, and development of structural changes including glomerular sclerosis, tubular atrophy, and interstitial fibrosis. Globotriaosylceramide (GL-3) deposition is probably the initiating factor of the disease pathology. The begining of enzyme replacement in 2001 for Europe and 2003 for the United States marked a turning point for the clinical management of Fabry patients. There are no studied to definitively guide timing of enzyme replacement therapy, and duration of therapy. As there is growing evidence that renal outcomes are more directly related to the degree of fibrosis and scarring, preventing the development of these irreversible changes by early initiation of enzyme replacement therapy may have the greatest impact on renal outcomes 關鍵字: 法布瑞氏症, 阿爾法半乳糖甘酶缺乏,蛋白尿 Key words: Fabry disease, galactosidase A deficiency, protainuria, globotriaosylceramide