CEM214 LESSON 4 (08)
advertisement
")
CEM214 LESSON 4 (08)
∆U FOR CHEMICAL REACTIONS
From the definition of H = U + PV, it follows that
∆U = ∆H - ∆(PV)
For liquids and solids: ∆(PV) ≈ 0 and ∆U ≈ ∆H
For gases ∆(PV) = ∆ngRT if T is constant
With ∆U = ∆H - ∆ngRT
∆U FROM AVERGE BOND ENERGIES FOR GASES
∆U ≈ (sum of all bond energies in reactants) – (sum of all bond
energies in products) NB: ∆U is an estimation
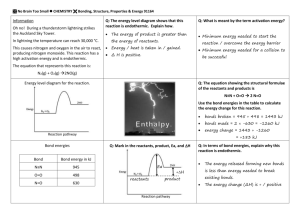
BOND ENTHALPY AND MEAN BOND ENTHALPY
Definition:
The standard molar enthalpy change of bond dissociation (Hd° is
the energy change when 1 mole of bonds is broken, the molecules
and resulting fragments being in the gaseous state at 298K and a
pressure of 100kPa (1 bar).
1
Introduction
This energy refers to a specific bond in a molecule, but if a
molecule has 4 of the same bond (eg the C-H bonds in
methane), then different dissociation energies can occur.
CH4(g) => CH3(g) + H(g)
Hd° = +427 kJ mol-1
CH3(g) => CH2(g) + H(g)
Hd° = +371 kJ mol-1
So it is much more useful to know the average amount of energy
needed to break a particular bond.
In this case, the process of breaking all the bonds in methane,
ending up with gaseous atoms.
So this process could be written as:
The enthalpy change for this reaction is +1646 kJ mol-1 , so the
average bond enthalpy is +1646 / 4 = +412 kJ mol-1 . They can be
looked up in data tables.
It is important to stress that these are mean or average bond
enthalpies. If Average bond enthalpies are used to calculate an
2
enthalpy change, the answer will be slightly out compared to a
result obtained by other methods.
Why are they useful?
Their main use is in working out enthalpy changes for reactions. If
we know the amount of energy needed to break a bond
(endothermic), and the amount of energy we get back when a new
bond forms (exothermic), then we can quite easily work out an
approximate (because of the average nature of the bond
enthalpies) value for the enthalpy change!
Remember: Breaking bonds requires energy (Endothermic)
while making bonds releases energy (Exothermic).
Don't be scared by this equation below, it is just the correct
terminology as used by chemists for working out enthalpy changes
from bond energies.
This basically means that you add up all the energies of the broken
bonds, add up all the energies of the bonds that are reformed and
subtract one from the other.
3
It’s another version of Hess' Law. Similar to the one that can be
used when you know all the enthalpies of formation for the
substances in a reaction.
An example.
The complete combustion of propane can be represented by the
following equation:
or we could redraw it to represent the bonds present:
We now need to work out how many of each bond type we have
broken.
8 x C-H
2 x C-C
5 x O=O
And then how many bonds have been formed!
6 x C=O
8 x H-O
4
So using data tables we can look up then average bond enthalpies
from, and calculate the enthalpy change of the reaction.
Bond Type
Average bond enthalpy /kJ
mol-1
C-H
+413
C-C
+347
O=O
+498
C=O
+805
H-O
+464
Notice they are all endothermic.
So we can now do the sum, remember, sum of bonds broken - sum
of bonds formed.
Hr°= [(8x413)+(2x347)+(5x498)] - [(6x805)+(8x464)]
= - 2054 kJ mol-1
The value for the enthalpy of combustion of propane from the data
table is -2219kJ mol-1 .
This apparent error is due to fact that we use Average Bond
Enthalpies in our calculations and the fact that the values above
relate to the gaseous state, while the standard combustion state of
5
water is liquid. If we allow for this, we get a value of -2226 kJ
mol-1 which is pretty close to the value obtained above. The
remaining difference must be down to the average bond enthalpy
factor.
Exercise
Using the average bond energy values, estimate ΔU for the
following reaction
C2H2(g) + 5/2 O2(g) → 2CO2(g) + H2O(g).
Solution
ΔU ≈ (sum of all bond energies in reactants) - (sum of all bond
energies in product)
ΔU ≈1mol x BE(C≡C) + 2 mol x BE(C-H) + 5/2 mol x BE(O=O)
– 2 mol x 2 x BE(C=O) – 1 mol x 2 x BE(H-O)
ΔU ≈1mol x 812 kJ mol-1 + 2 mol x 416 kJ mol-1 + 5/2 mol x
498 kJ mol-1 – 2 mol x 2 x 799 kJ mol-1 – 1 mol x 2 x 464 kJ
mol-1
ΔU ≈ 2889 kJ – 4124 kJ = -1235 kJ
1. Estimate the combustion of CH3OH(l) by using the appropriate
average bond energies data. Assume that the evaporation enthalpy
of CH3OH(l) = 38 kJ mol-1 and that of H2O(l) = 44 kJ mol-1.
6
Bond Energies
Since we are treating the chemical bond as largely depending only
upon the nature of the two atoms in contact through the bond,
perhaps we can use this idea to determine the overall stability of a
molecule by adding up its bond energies. This assumes that all
chemical bonds between the same pair of atoms of the same
type are approximately equal in properties. Namely, in this
case, we will assume all C-H bonds take about the same amount of
energy to break, regardless of the molecule they are in.
(3) Bond Dissociation enthalpy (bond energies)
7
The bond dissociation enthalpy (bond energy) represents the
energy required to break one mole of chemical bonds in the gas
phase. The term 'mean bond dissociation energy' is more
commonly used, since the actual energy required to break a
particular bond is dependent on the precise environment of the
bond.
Consider the dissociation of methane by successive breakage of CH bonds:
We have:
(i)
(ii)
(iii)
(iv)
Given this information we may estimate the mean bond
dissociation enthalpy for C-H given by:
8
CEM214 LESSON 5
IDEAL GAS RELATIONSHIPS
Reasons to devoting careful study to ideal gases:
1. Ideal gases are the simplest systems to deal with and
they therefore provide us with valuable and not too
difficult exercises for testing our understanding of the
subject.
2. Some of the simplest conclusions that we draw for ideal
gases can readily be adapted to more complicated
systems such as solutions.
REVERSIBLE COMPRESSION AT CONSTANT PRESSURE
Consider reducing the volume of an ideal gas by lowering its
temperature at constant pressure.
We need to find:
How much work is done on the system during this process
How much heat is lost and
What the changes are in internal energy and enthalpy
Suppose we have 1 mol of an ideal gas confined in a cylinder with
a piston, at a pressure P1, a molar volume Vm,1 and an absolute
temperature T1.
9
See fig. 2.6 (a) for the isotherm (i.e., PV relationship) at this
temperature. Appendix 1(p75)
Point A → represents the initial state. Isotherm 1 ~ T1.
Point B → heat is removed from the system reversibly at constant
pressure P1 until the volume has fallen to Vm,2. This can be
achieved by lowering the temperature of the system by an
infinitesimal amounts to T2.
Isotherm 2 ~ T2.
Work done on the system w:
Vm, 2
Wrev = - P dV = P1 (Vm,1 – Vm,2)
1
2.54
Vm,1
NB: This is true whether the gas is ideal or not.
If the gas is ideal, use of the gas law PVm = RT (1 mol) leads to
the expression:
W rev.
= P1 {(RT1/P1) – (RT2/P1)}
= R(T1 – T2)
(ideal gas only)
2.56
10
Since V1 > V2 and T1>T2
Positive work is done ON the system. (The shaded area in Fig
2.6(a)
If the system had expanded isothermally and at constant pressure,
the shaded area would represent the work done BY the system.
The heat absorbed / released by the system during the process
A→B
is given by:
T2
qp,m = c p dT
T1
= Cp,m (T2-T1)
2.58
= ∆Hm (Enthalpy)
2.59
Since T1 > T2, -qP,m released by the system = -∆Hm
The molar internal energy change ∆Um (also negative for this
process) is obtained by the use of the first law:
∆Um = q + w = Cp,m (T2-T1) + R(T1 – T2)
= (Cp,m – R) (T2-T1) = CV,m (T2-T1)
Since CV,m + R = Cp,m
2.60
2.61
from
2.36
= Cp,m (T2-T1) - R (T2-T1) = CV,m (T2-T1)
∆Hm - ∆(PV) = ∆Um
11
Rearranging:
∆Hm = ∆Um + ∆(PV)
2.62
REVERSIBLE PRESSURE CHANGE AT CONSTANT
VOLUME
Consider 1 mol ideal gas if initial state of P1,Vm,1, T1 to the final
state P2, V m,1, T2.
See fig. 2.6 (b) :
P1 is higher than P2. To accomplish this we must remove heat
infinitesimally from T1 to T2. (Reversibly)
The work done on the system w = area below the line AC in the
fig. 2.6(b). = zero
To confirm:
Vm, 2
Wrev = - P dV = 0
1
2.63
Vm,1
Since the process occurs at constant volume.
12
The heat absorbed is given by
T2
qV,m = cV ,m dT = CV,m (T2-T1)
2.64
T1
since (T2 < T1) the expression CV,m (T2-T1) is negative
CV,m (T2-T1) = ∆Um.
2.65
The value of ∆Hm is obtained as follows:
∆Hm = ∆Um + ∆(PV) = ∆Um + ∆(RT)
2.66
= CV,m (T2-T1) + R(T2-T1)
= (CV,m + R)(T2-T1)
∆H = Cp,m (T2-T1)
2.67
2.68
Interesting to compare the two processes
A → B on fig. 2.6 (a) and
A → C on fig. 2.6 (b)
Work and heat values are different in the two cases
However, the ∆H and ∆U values are the same.
This implies that ∆U internal energy is the same at point B on
the T2 isotherm as it is at point C.
The same is true for the enthalpy ∆H.
This can be proved for any two points on an isotherm
In Conclusion: THE INTERNAL ENERGY AND ENTHALPY OF AN
IDEAL GAS DEPEND ONLY ON THE TEMPERATURE AND REMAIN
CONSTANT UNDER ISOTHERMAL CONDITIONS
13
REVERSIBLE ISOTHERMAL COMPRESSION
Another process of great importance: The compression of an ideal
gas along an isotherm (i.e., at constant temperature, T)
Illustration in Fig.2.6 ( c)
The initial conditions are: P1,Vm,1, to the final state P2, Vm,2, with
Vm,1> Vm,2.
Remember that for an isothermal process
∆Hm = 0 and ∆Um = 0
The work done on the system in a reversible compression is
Vm, 2
Wrev = - P dV
Vm,1
1
Because P is varying, we must express it in terms of Vm by use of
the ideal gas equation: for 1 mol PVm = RT;
Thus
Vm, 2
Wrev = -
Vm,1
RT
dV
V
= -RT ln V│Vm.1Vm,2
= -RT ln Vm,2/ Vm,1
2.71
Since Vm,1 > Vm,2this is a positive quantity
14
Wrev = RT ln Vm,1/ Vm,2
2.72
The heat absorbed is found from the first law:
∆Um = qrev + wrev
2.73
qrev = ∆Um - wrev = 0 - wrev = RT ln Vm,2/ Vm,1
2.74
This is negative. Heat is evolved during compression.
NB: When we compress a gas, we do work on it and supply
energy to it; if the temperature is to remain constant, heat must be
evolved.
Using concentrations to measure work done
If 1 mol of gas has a volume Vm,1 the concentration is
C1 = 1/Vm,1
Similarly,
C2 = 1/Vm,2
The ratio of volumes is therefore the inverse ratio of the
concentrations:
Vm,2 / Vm,1 = c1/c2
Equation 2.72 for the work done in the isothermal reversible
expansion of 1 mol of an ideal gas can be written alternatively as
wrev,m = RT ln c2/c1
For n mol,
wrev,m = nRT ln c2/c1
Examples 2.8 and 2.9
15
REVERSIBLE ADIABATIC COMPRESSION
Final process: Consider the compression of an ideal gas contained
in a vessel whose walls are perfectly insulating, so that no heat can
pass through them
Such a process is called ADIABATIC.
See Fig 2.6 (d)
NB
Work is done on the gas in order to compress it
No heat can leave the system : dq = 0
The final temperature T2 must be higher than the initial
temperature T1.
The Fig shows the two T1 and T2 isotherms as well as the adiabatic
curve AB.
Consider n mol of the ideal gas.
According to the first law: dU = dq – PdV
16
dU + PdV = 0 = dq
for n mol, dU = nCV,m dT + PdV = 0
2.82
This is true whether the gas is ideal or not.
For n mol of an ideal gas PV = nRT
2.83
CV,m dT/T + R dV/V = 0
2.84
Integrating between temperatures T1 and T2 and the volumes V1
and V2 assuming that CV,m is a constant.
CV,m ln T2 / T1 + R ln V2 / V1 = 0
So ln T2 /T1 + (Cp,m - CV,m) / CV,m ln V2 /V1 = 0
The ratio Cp,m to CV,m is often written as γ
γ = Cp,m / CV,m
ln T2 /T1 = (γ – 1) ln V2 / V1 = 0
or
T2 / T1 = {V1 / V2} γ – 1
2.90
Conclusion
U and H remain unchanged along the isothermal T1 also for
isothermal T2.
∆U = Cv(T2 –T1)
And
∆H = Cp(T2 - T1)
Since ∆U = q + w, and q = 0
w= Cv(T2 – T1) = nCv,m(T2 – T1)
2.97
17
For ideal gases the following conditions hold:
1. PV = nRT
U
2. V
T
=0
For non ideal gases, neither of these conditions is satisfied.
Study section 2.7.
1. A system of 5 mol of an ideal gas is compressed isothermally at
300 K from 25 L to 5 L under reversible conditions. Calculate
i. the work done w
ii. the change in internal energy ∆U
iii. The change in pressure ∆P.
2.
18