Supplementary table 1: SPG42 sequencing and HRM
advertisement

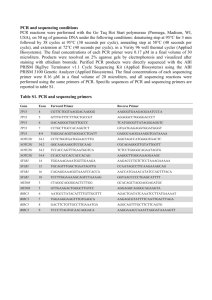
Schlipf et al., supplement, page 1 220 patients with autosomal dominant spastic paraplegia do not display mutations in the SLC33A1-gene (SPG42) Nina A. Schlipf, Christian Beetz, Rebecca Schüle, Giovanni Stevanin, Anne Kjersti Erichsen, Sylvie Forlani, Cécile Zaros, Kathrin Karle, Stephan Klebe, Sven Klimpe, Alexandra Durr, Susanne Otto, Chantal ME Tallaksen, Olaf Riess, Alexis Brice, Peter Bauer and Ludger Schöls Supplementary Material SPG42 amplification and high resolution melting (HRM) analysis The primers used to screen the SPG42 gene SLC33A1 (ENSG00000169359) were designed to flank the coding regions of SPG42 exons 1-6 and to amplify fragments of an average size of 259 bp (ranging from 206 to 298). All primers were 5´tailed with universal M13 or revM13 sequences to facilitate sequencing. Primer sequences are available in supplementary table1. The PCR and HRM was performed in a single run on a LightCycler®480 instrument (Roche Diagnostics, Mannheim, Germany) with a total reaction volume of 10 µl in a 384-well microtiter plate. The reaction mixture containing 20ng of genomic DNA, 2.5 mM MgCl2 (Roche Diagnostics, Mannheim, Germany), 10 pmol of each primer, 5 µl 2x LightCycler®480 High Resolution Master Mix (Roche Diagnostics, Mannheim, Germany) and PCR grade water in a volume of 10 µl. The PCR cycling conditions included an activation step at 95°C for 10 minutes followed by 45 cycles of 95°C for 10 seconds, a touch down of 68°C to 58°C for 15 seconds (1°C/cycle) and 72°C for 20 seconds. The amplicons were denaturated at 95°C for 1 Schlipf et al., supplement, page 2 minute, renaturated at 40°C for 1 minute. Afterwards the HRM was executed over the range from 65°C to 95°C with 25 signal acquisitions per degree. The HRM analysis was performed using Gene Scanning Software Version 1.5.0 (Roche Diagnostics, Mannheim, Germany), which allows clustering the samples into groups on the basis of difference plot obtained analyzing the differences in melting curve shapes. To guarantee a high detection sensitivity, HRM analysis sensitivity settings were 0.7 (high sensitivity). Schlipf et al., supplement, page 3 Supplementary table 1: SPG42 HRM amplification and sequencing primers Exon SPG42 Exon01 Fragment Lenght primer sequence 5´->3 250 bp 1-1F 1-1R 259 bp 1-2F 1-2R 277 bp 1-3F 1-3R 239 bp 2-1F 2-1R 246 bp 2-2F 2-2R M13-CGGATCCAAAACGCTCAG revM13GGCTTTTAAGAAGTCGCCAGT M13-AGGCGGTTGGGATGACAGT revM13GGGCCAAAAGACAAAACTGA M13-AAGCATCCCACTCATTTTGC revM13CACACACTCTTTCAAAAGGATAAA T M13-GCATGAGCACCTGTACTTGG revM13TGTGATCCCTTGTGTTTCTTCTT M13-ATTGGTTGCCCTTCTGAAAA revM13TGGTAGTGGAAAGGGACCTG M13TGCATGGAGGAAAATATTTCATAA revM13TGTTCCTCATTTTACCCTTTCTT M13GCTAAAATGGAAGTCAACTCAATG revM13TCTTCAGTGTCAGATTCATGCTT M13TTTCAGGTTTATGACTTTGATGTTC revM13-TTTCCTCCCAGATTGGACAC M13CAATGCAAAGGTTAGTGATCCA revM13CCAGAACCTATGGACAGATATGA M13TTTCATTTTGAAATTGTGGAGTATT revM13CCTTGCTAGAATGTCCAGTAGCA SPG42 Exon02 3F SPG42 Exon03 298 bp 3R 4F SPG42 Exon04 277 bp 4R 5-1F 206 bp 5-1R 5-2F SPG42 Exon05 294 bp 5-2R 6F SPG42 Exon06 244 bp 6R Schlipf et al., supplement, page 4 Direct sequencing To re-amplify the amplicons, we used the same primers as for HRM (see supplementary table 1). PCR was performed in 20 µl volumes. Amplification reactions included 10ng genomic DNA, 10 pmol of each primers, 10 mM of dNTPs (Fermentas GmbH, St. Leon-Rot, Germany), 5x PCR buffer (including 25 mM MgCl2) (Promega GmbH, Mannheim, Germany), 0.5U Go Taq (Promega GmbH, Mannheim, Germany) DNA Polymerase and H2O to a volume of 20 µl. All 10 Amplicons were amplified using a touch down protocol. The cycling conditions which were run on a DNA Engine Dyad thermal cycler (Bio-Rad Laboratories, München, Germany), consisted of an initial denaturation step at 94°C for 3 minutes, followed by 8 cycles of denaturation at 94°C for 30 seconds, annealing starting at 68°C for 30 seconds (decreasing 1°C/cycle) and extension at 72°C for 60 seconds. This step was followed by 27 additional cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 30 seconds and extension at 72°C for further 90 seconds. Final extension was carried out at 72°C for 2 minutes. The PCR products were purified using ethanol precipitation according to the instructions of the manufacturers. The purified PCR products were eluted in 15 µl volume and then used as template in cycle sequencing using Big Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems Inc, Foster City, CA, USA). The reaction mix consisted of 2 µl ABI BigDye 3.1, 10 pmol primer, 1 µl ABI BigDye Buffer and approx. 20ng purified PCR template in a 10 µl total volume. The sequencing reactions were run on a DNA Engine Dyad thermal cycler (Bio-Rad Laboratories, München, Germany) according to the following protocol; one cycle of 94°C for 1 minute; 30 cycles of 94°C for 10 seconds, 50°C annealing temperature for 5 seconds, 60°C for 4 minutes. The sequencing reaction was purified using ethanol precipitation Schlipf et al., supplement, page 5 and run on an ABI3100 Genetic Analyzer (Applied Biosystems Inc, Foster City, CA, USA). All sequences were examined using SequencePilot Software (JSI medical systems GmbH, Kippenheim, Germany).