Role of cyclooxygenase-2 in tumor progression and immune
advertisement

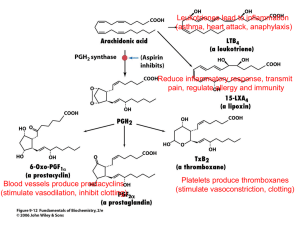
Indian Journal of Biochemistry & Biophysics Vol. 44, December 2007, pp. 419-428 Minireview Role of Cyclooxygenase-2 in Tumor Progression and Immune Regulation in Lung Cancer Swati Patel and Shubhada Chiplunkar Chiplunkar Lab, Advanced Centre for Treatment, Research and Education in Cancer (ACTREC), Tata Memorial Centre, Kharghar, Navi Mumbai 410 210, India Received 29 August 2007; revised 08 October 2007 Lung cancer is the leading cause of cancer death all over the world. The low 5-year survival rate (under 15%) has changed minimally in the last 25 years. Amongst different types of lung cancers, non-small cell lung carcinoma (NSCLC) types account 25-40%. To improve the survival of lung cancer patients, new therapeutic strategies are needed. The search for improved therapies has led to the investigation of agents that target novel pathways involved in tumor proliferation, invasion, survival and immune regulation. Cyclooxygenase-2 (COX-2) is one of the novel targets under evaluation for NSCLC therapy and chemoprevention. Although multiple genetic alterations are necessary for lung cancer invasion and metastasis, COX-2 may act as central element in orchestring these processes. COX-2 plays an important role in all aspects of tumor development and growth. It also plays a pivotal role in regulation of cytokines and immune responses in NSCLC patients. In this article, we review the experimental and clinical evidences on the possible link between COX and NSCLC. Keywords: Cyclooxygenase (COX), Non-small cell lung carcinoma (NSCLC), Non-steroidal anti-inflammatory drugs (NSAIDs) Introduction Lung cancer results in an enormous clinical burden for health-care providers with very high mortality rates. Despite extensive research, the overall 5-year survival rate is only 8-14% and has improved only marginally during the past 25 years. Amongst different types of lung cancers, non-small cell lung carcinoma (NSCLC) types account 25-40%. To improve the survival, advances are needed in early __________ For correspondence Tel.: +91-022-27405032; Fax: +91-022-27412894 E mail: schiplunkar@actrec.gov.in Abbreviations: AA, arachidonic acid; APC, antigen presenting cell; Bcl-2, B-cell lymphoma leukemia-2; C/EBP, CCAAT enhancer-binding protein; DCs, dendritic cells; ECM, extracellular matrix; EGF, epidermal growth factor; EGFR, EGF receptor; EP, PGE2 receptor; ERK, extracellular signal-regulated kinase; GSK-3b, glycogen synthase kinase-3b; IP3, inositol trisphosphate; JNK, c-Jun N-terminal kinase; LAT, linker for activation of T cells; LEF, lymphoid enhancer factor LPS, lippopolysaccharide; MAPK, mitogen-activated protein kinase; MEK1/2, mitogen-activated extracellular signal-regulated kinase; NSCLC, non-small cell lung carcinoma; 15-PDGH, 15-hydroxyprostaglandin dehydrogenase; PGs, prostaglandins; mPGES, microsomal prostaglandin E synthase; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PLA2, phospholipase A2; PLC, phospholipase C; PPAR, peroxisome-activated receptor; Src, nonreceptor tyrosine kinase; TCF, T cell factor; TNM, tumors, nodes and metastases. diagnosis and in development of new therapeutic strategies. These require a better understanding of the molecular mechanisms underlying cell transformation and tumor progression. New therapies could target molecular pathways essential for tumor proliferation, angiogenesis, apoptosis and immune suppression. Cyclooxygenase (COX)-2 is one of the novel targets under evaluation for NSCLC therapy and chemoprevention. It plays an important role in all aspects of tumor development and growth. Evidences suggest that it is involved in multiple pathways, conferring the malignant and metastatic phenotype in lung cancer. Although multiple genetic alterations are necessary for lung cancer invasion and metastasis, COX-2 may act as central element in orchestring these processes1-3. Various studies have shown association of overexpressed COX-2 with apoptosis resistance4,5, angiogenesis6, decreased host immunity7-9 and enhanced invasion and metastasis10,11. In this article, we have reviewed experimental and clinical evidences on the possible link between COX and NSCLC with respect to tumor progression and immunoregulation. Biochemistry of COX-1 and COX-2 Receptor activation or cell injury signals causes release of arachidonic acid (AA) from lipid membrane 420 INDIAN J. BIOCHEM. BIOPHYS., VOL. 44, DECEMBER 2007 stores by activity of lipases, such as cytosolic phospholipase A212. COX catalyzes the cyclization of AA to PGH2, hence also known as prostaglandin H synthase (PGHS)13. It accomplishes this by first generating prostaglandin (PG) G2, by conversion of polyunsaturated 20-carbon AA to varying degree of unsaturation by oxidative cyclization of central 5 carbon atoms. COXs also have peroxidase activity and catalyze hydroxylation of cyclopentens through endoperoxidation at position 15C converting PGG2 to PGH2. Isomerase enzymes that are expressed in both tissue and cell type selective manner govern the next step, namely conversion of PGH2 to cell type specific production of individual prostanoids. Five primary prostanoids PGE2, PGD2, PGF2, PGI2 and tromboxane A2 (TXA2) are produced by different cell types which are involved in different physiological functions (Fig. 1). Two isoforms of COX, COX-1 and -2 are known COX-1 is constitutively expressed in most cells and tissues and regulates normal house-keeping functions14, while COX-2 is inducible in response to various stimuli like cytokines and growth factors in most cells, except in neuronal and renal cells in which it is constitutively expressed14. Despite overall similarities, two enzymes show subtle differences in the amino acid composition at the active sites. COX-2 has valine at positions 89 and 523, while COX-1 has isoleucine, resulting in large space availability in the former. Further, the presence of valine at position 434 in COX-2 as against isoleucine in COX-1 allows a gate mechanism to operate in favor of the former15. These differences in active site structures provide specificity to COX inhibitors as selective COX-1 or COX-2 inhibitors. Also, despite overall similarities in structure, substrate and catalytic activities, both the isoforms display differences in their expression profiles and in the panel of cellular responses evoked. These activities of COX are likely to be dictated by a number of factors, including intracellular localization and coupling to specific prostaglandin synthases, as well as by the specific cellular pattern of expression of prostanoid receptors, which are known to trigger different intracellular signaling pathways. The major metabolite of COX pathway is PGE216, which regulates key responses in humans including reproductive, gastrointestinal, neuroendocrine and immune system. PGE2 exerts its effect through G protein coupled 7 span transmembrane receptors EP1-4. Fig. 1 — PGE2 biosynthesis pathway [Phospholipase A2 (PLA2) catalyzes conversion of phospholipids to arachidonic acid which is converted to prostaglandin G2 (PGG2) by COX-1/2, which in turn gets converted to prostaglandin H2 (PGH2) by peroxidase activity of COX isoforms. PGH2 is then converted to prostaglandin I2 (PGI2), thromboxane A2 (TxA2), prostaglandin E2 (PGE2), prostaglandin D2 (PGD2) and prostaglandin F2 (PGF2) by specific isomerases and synthases depending upon type of tissue or cells involved. PG, prostaglandin, Tx, thromboxane] COX-2 in NSCLC Patients COX-2 overexpression and high production of PGE2 is reported in variety of malignancies including NSCLC1,3,7. COX-2 activity has been detected throughout the progression of a premalignant lesion to the metastatic phenotype. Higher COX-2 expression has been observed in lung cancer lymph node metastasis, compared to primary adenocarcinoma1. COX-2 over expression appears to portend a shorter survival among patients with early stage NSCLC and the extent of its expression is found to be associated with both decreased overall and diminished diseasefree survival rates17. In another study, linear relationship between COX-2 expression and aggressive behavior of tumors in NSCLC and increased mortality rate has been found18. Association of COX-2 expression with poor prognosis, independent of TNM staging in surgically-resected NSCLC patients has also been reported19. A polymorphism in the COX-2 gene with an increased risk of lung cancer has been observed20. Thus, these reports suggest the involvement of COX-2 in the pathogenesis of NSCLC. COX-2 in Tumor Progression Smoking and other mutagenic environmental factors capable of inducing cell transformation play PATEL & CHIPLUNKAR: COX-2 IN LUNG CANCER an important role in tumor progression in the respiratory tract. COX-2 activates tobacco smoke carcinogens such as benzoapyrene (BaP)21. It catalyzes the conversion of BaP-7, 8-dihydrodiol to BaP-diol epoxide, which binds to DNA22. Also, BaP has been demonstrated to induce epithelial COX-2 expression and PGE2 production23. COX-2 expression is induced by the various inflammatory stimuli such as TGF-β, IL-1β, hypoxia and epidermal growth factor (EGF) generated in pulmonary microenvironment of those at high risk of lung cancer24. In addition, various types of mutations and oncogenes expressions are also associated with high COX-2 expression and progression of lung tumors. Wild-type tumor suppressor p53 suppresses COX-2 transcription, but not the mutant p53, suggesting that p53 mutation also has a role in COX-2 overexpression25. Also, K ras and β catenin mutations are associated with elevated COX-2 expression26. CCAAT/enhancer binding protein (C/EBP) and activating transcription factor/cAMP response element binding protein (ATF/CREB) also play a critical role in the regulation of basal COX-2 expression in murine lung carcinoma27. These molecules can be potential target for down regulating COX-2 expression in lung cancer. Transcription regulation of COX-2 also involves nuclear factor of k chain in B cell (NF-kB), activator protein-1 (AP-1) and nuclear factor of activated T cells (NF-AT). COX-2-/- mice have shown attenuation of Lewis lung carcinoma (LLC) tumor growth, but not the wild type and COX-1-/- mice, confirming the role of COX-2 in tumor progression28. Microtubule-interfering agents such as taxol can stimulate COX-2 transcription via ERK and p38 MAP kinase pathways and promotes carcinogenesis by overexpressing COX-229. COX-2 in Angiogenesis Angiogenesis is an important factor in tumor development. Tumor associated angiogenesis is mediated by the migration and proliferation of host endothelial cells and is a requisite for tumor growth30. The mechanisms for promotion of tumor-associated angiogenesis are suggested to be activated in the early stages of tumor development31. Vascular endothelial growth factor (VEGF), transforming growth factor (TGF) and β, basic fibroblast growth factors (FGF) and chemokines like IL-8 are implicated in tumorrelated angiogenesis in lung cancer32. 421 Because COX-2 has been reported to be localized in both tumor cells and the adjacent stromal cells, PGs generated through the presence of COX-2 might act on the tumor cells (a cell autonomous effect) or surrounding stroma cells (a cell non autonomous or landscaping effect) to facilitate tumor development33. Thus, the products of COX pathway in tumor milieu may promote tumor growth. PGE2 not only activates G coupled proteins, but also indirectly activates Wnt signaling, peroxisome poliferator activated receptors (PPAR) and epidermal growth factor receptor (EGFR) pathway34. It also up regulates Wnt target genes, such as cyclin D1 and VEGF35. It is a potent vasodilator and can act synergistically with other mediators and chemotaxins to increase microvascular permeability35. In conjunction with cytokines, PGE2 is a central mediator of febrile responses36. Genetic mutations of various oncogenes and tumor suppressor genes and dysregulated immune responses are associated with regulation of these angiogenic mediators37. Host-derived COX-2 products are shown to be instrumental for sustained tumor growth, as lung cancer cells injected into syngeneic mice bearing a genetic background of COX-2 (not COX-1) deficiency have shown a markedly reduced growth rate28. Expression of COX-2 and nitric oxide synthase (NOS)-2 is correlated with the VEGF expression level as well as microvascular density in tumors of NSCLC patients38. COX-2-dependent expression of ELR+ angiogenic chemokines CXCL5 and CXCL8 has been demonstrated in vitro and in vivo in NSCLC. In addition, CXCL5 and CXCL8 are decreased with inhibition of COX-239. Treatment with anti-CXCL5 or anti-CXCL8 neutralizing antibodies results in a significant decrease in tumor growth. Moreover, neutralization of PGE2 leads to a decrease in CXCL5 and CXCL8 production39. This implies specific role of PGE2 in angiogenesis and neovasculararization during tumor development process. COX-2 in Tumor Invasion and Metastasis COX-2 can also affect cell invasion, which is vital for the dissemination of metastatic cells across extracellular matrix (ECM) and spread to distant organ sites. Its elevated expression has been shown to increase tumor invasiveness and enhance metastatic potential28,40. Lung cancer cells with elevated COX-2 also display concomitant increase in expression of CD44, a cell surface receptor for hyaluronate, a major 422 INDIAN J. BIOCHEM. BIOPHYS., VOL. 44, DECEMBER 2007 glycosaminoglycan component of ECM41. In fact, antibody-mediated blockade of COX-2-derived PGE2 is sufficient to decrease CD44 and matrix metalloproteinase-2 (MMP-2) expression as well as invasion in an EP4-dependent manner9. Exposure of NSCLC cells to PGE2 up-regulates CD44, EP4, and MMP-2 expression and enhances cell invasion. CD44 induces co-clustering with MMPs and can, therefore, promote MMP activity, tumor invasion and angiogenesis42. In monocytes, PGE2 is found to activate membrane type-1 MMP (MT-1 MMP) and thereby promotes activation of MMP-243. This study in NSCLC indicates an important autocrine/paracrine role for PGE2 in regulation of CD44 and MMP-2dependent invasion in human NSCLC. Laminin 5, an ECM protein involved in cell migration and invasion is frequently expressed in several different malignancies10. It is also coexpressed with COX-2 in early stage lung adenocarcinomas10. EGFR signaling pathway has been shown to induce both COX-244 as well as laminin 510. Also, RAS mutations in multiple myeloma cells cause selective induction of COX-2, resulting in high binding to ECM protein and chemotherapeutic drug resistance, conferring high metastatic properties to these cells45. Another mechanism of metastasis and invasion in lung cancer includes lymphangiogenesis or formation of new lymphatic vessels46,47. Another study has demonstrated that human lung adenocarcinoma cells that overexpress COX-2 significantly increases vegf-c gene expression, associated commonly with lymphangiogenesis in animal models11. Induction of vegf-c occurs through PGE2 activation of the Her2/neu tyrosine kinase signaling pathway via the EP1 receptor. Thus, this study provides further evidence for interactions between COX-2 and human EGF signaling in the development and progression of lung cancer. COX-2 in Apoptosis Early tumor growth is dependent on the balance between the rates at which cells are produced through cell division and die through a natural cell death process known as programmed cell death or apoptosis. Defects in apoptosis pathway allow tumor cells to survive for prolonged periods of time, accumulate genetic errors and live in a suspended state that permits metastatic spread. Also defects in apoptosis contribute to resistance to radiation or chemotherapy4,5,48. In hostile and often hypoxic tumor environment, genetic mutations may allow cells to develop resistance to apoptosis and increased metastatic potential4,5. Studies have demonstrated the role of COX-2 in this process48. Increased cell survival has been observed in lung cancer cells expressing high COX-248. Selective COX-2 inhibitors have been shown to induce apoptosis in lung carcinoma cells48. This ability to induce apoptosis is of particular interest, considering their potential with other treatment modalities such as radiation therapy. Preclinical studies have indicated improved response on combined treatment with celecoxib (a COX-2 inhibitor) and radiation therapy. Studies have shown that the Ras/Raf/MAPK signaling pathway is necessary for transcriptional induction of COX-2 by several kinds of stimuli and COX-2 is linked to epithelial tumor cell survival29,49. Apoptosis induction has been widely investigated to define the potential anti-neoplastic mechanisms of COX-2 inhibition. In the landmark study, forced expression of COX-2 has been found to increase Bcl2 expression and resistance to apoptosis4. Subsequent studies in a variety of tumors suggest that both Bcl-2dependent50 and independent5 pathways may be operative. Selective COX-2 inhibitors also induce apoptosis in several different types of tumors like prostate, colorectal and colon50,51, including lung cancer48,52. The overexpression of COX-2 or exposure to PGE2 can increase apoptosis threshold in lung adenocarcinoma cells by up-regulation of the mcl-1 gene in a PI3K/Akt-dependent manner18. A strong correlation has also been found between expressions of anti-apoptotic protein survivin and COX-253. High tumor COX-2 expression leads to decreased ubiquitination and stabilization of survivin, an effect replicated with exogenous PGE2 treatment and inhibited by COX-2 inhibitor. Also, PGE2 rapidly stimulates ERK phosphorylation and proliferation in a subset of NSCLC cell lines, contributing to increased migration and resistance to EGFR inhibitors53. COX-1 and 2 in Immune Regulation The immune system is able to recognize tumor cells and eliminate them54. Paradoxically, tumor cells acquire various characteristics, which allow them to evade this immunological surveillance through ‘‘tumor escape mechanisms’’. The spreading of cancerous growth is also dependent on successful evasion of the immune system. Chronic inflammation is one such process, which helps in tumor growth and PATEL & CHIPLUNKAR: COX-2 IN LUNG CANCER progression55. A key role for COX-2 in inflammation has been established56. Various studies have also shown involvement of COXs in immune regulation and surveillance56-60. COX-1 and 2 in T cell Development COX-1 and 2 are expressed in T lymphocytes and appear to play a role in T cell development, activation, chemotaxis, polarization and apoptosis57-60. PGE2 is also found to modulate cellular proliferation of T cells in thymus59. In thymus, COX-1 is expressed in developing (CD4-CD8- and CD4+CD8+) thymocytes, whereas COX-2 in a subset of medullary stromal cells59. Both isoforms play a distinct role in thymic development. COX-1 (not COX-2) is present in immature thymocytes, whereas COX-2 expression changes as the thymus matures. COX-1-dependent formation of PGE2 acting through EP2 receptors plays an important role in double-negative to doublepositive transition. COX-2 expression in the stroma influences positive selection of thymocytes through EP1 receptors in CD4 single positive formation59. Mice deficient for COX-1 show an early block in thymic development, resulting in a significant reduction in double-positive thymocytes, whereas COX-2 deficiency selectively affects maturation of single-positive CD4+ thymocytes59. Phenocopies of COX deficiency can be obtained by pharmacologic inhibition of specific COX isozymes in the fetal thymus, supporting a crucial physiologic role of COX in T-cell development59. COX-1 and 2 in Activation and Functions of T Cells Activation of T cells through the T cell antigen receptor (TCR)/CD3 complex triggers a program of gene expression that can be divided into three phases — immediate events (independent on new protein synthesis), early events (dependent on protein synthesis and preceding cell division) and late events (occurring after cell proliferation)61. COX-1 and 2 play an important role in all these phases of T cell activation and cytokine production (Fig. 2). COX-1 is constitutively expressed in T cells, where as COX-2 is expressed as an early response gene, following TCR engagement through two NFAT motifs present in its promoter region, suggesting its role in T-cell activation62. This is further supported by the inhibitory activity of selective COX-2 NSAIDs on T-cell activation and cytokine production, which correlates with the inhibition of key transcription factors62. 423 COX-1 is required for TCR-dependent activation of p38 kinase, a member of mitogen-activated protein (MAP) kinase family activated in response to TCR engagement and required for T-cell activation and differentiation. It plays a pivotal role in Fyndependent activation of Rac and stress kinases. Thus, identifies COX-1 as an important early modulator of TCR signaling upon TCR engagement. Furthermore, COX-1 inhibition by selective NSAIDs results in impaired COX-2 expression, suggesting a sequential role of COX-1 and 2 in T-cell activation63. Selective COX-2 inhibitors have been found to negatively regulate proliferation, cell surface expression of activation markers (CD69, CD71, CD25) and cytokine production (TNF-, IL-2, and IFN-) by activated T cells62. These effects could be attributed to down-regulation of the transcription of genes such as NF-B and NF-AT by inhibition of COX-2, suggesting a role of COX-2 in tyrosine kinase signaling involved in activation of NF-B and NF-AT62. COX-2 in Maturation of Dendritic Cells Dendritic cells (DCs) are professional antigen presenting cells (APC) that are pivotal participants in the initiation of T-cell responses64. Activation of naïve T cells and their polarization into effector cells require interactions with mature DCs in the lymphoid organs. DCs acquire antigen in the periphery and subsequently transport it to lymphoid organs, where they prime specific immune responses64. Induction of COX-2 and PGE2 production via p38 MAPK is an early event observed even in APC upon lipopolysaccharide (LPS) stimulation65. PGD2, another product of COX-2 also partially inhibits the maturation of human monocyte-derived DCs through DP (receptor for PGD2) dependent or independent manner to favor the development of Th2 cells66. Collectively, it can be concluded that COX-1 and 2 are important immune modulators in development and functional regulation of immune responses, hence particularly playing a major role in pathogenesis of various diseases including NSCLC. COX-2 in Regulation of Immunity Tissue trauma normally causes infiltration of inflammatory cells into tissue and production of variety of cytokines or growth factors, suppressing or promoting cellular proliferation. If inflammation becomes persistent, the cytokine cascade that evolves could mediate either augmentation or suppression of 424 INDIAN J. BIOCHEM. BIOPHYS., VOL. 44, DECEMBER 2007 Fig. 2 — Role of COX-1 and COX-2 in T cell activation [Upon TCR activation, induction of proximal signaling events in lymphocytes requires constitutive activity of COX-1 to activate Fyn and ROS. Induction of these events along with other molecular pathways stimulates complex TCR activation cascade, transactivating bunch of transcription factors involved in COX-2 induction, proliferation and cytokine production. The whole cascade is critically regulated at each step. High PGE2 due to overexpression of COX-2, prolonged activation of G protein coupled receptor causes sustain ROS and Ras activation. This may lead to alteration of cytokine balance and lymphocyte response playing important role in pathogenesis of various diseases including malignancies] local immune responses, depending on the cellular make-up of the microenvironment. When transformed cells, interacting with inflammatory cells and growth factors (e.g., TNF-α) at sites of chronic inflammation continue to proliferate, the persistent inflammatory process may become a crucial step in carcinogenesis. Many cancers have been associated with persistent inflammation in lung carcinomas with asbestosis or silicosis67. These tumors directly interfere with the host immune system and release factors that modulate functions of immune cells or induce apoptosis of these cells. Lung cancer cells elaborate immunosuppressive mediators including type-2 cytokines, PGE2 and transforming growth factor-β (TGF-β) that may interfere directly with cellmediated, anti-tumor immune responses7. In addition to producing their own suppressive factors, tumor cells may also direct surrounding inflammatory cells to release suppressive cytokines in the tumor milieu68. Tumor-derived PGE2 is one mediator that orchestrates an imbalance in the production of suppressive and immune potentiating cytokines by lymphocytes and macrophages in the tumor environment7,8. COX-2 in Cytokine Regulation in NSCLC Expression of cytokines by T lymphocytes is a highly balanced process, involving stimulatory and inhibitory signaling pathway. The cytokine profile determines the effector functions of T cells. 3’,5’cAMP has long been recognized as a secondary messenger in the control of cellular proliferation69. cAMP elevating agents are known to induce IL-1070 and inhibit IL-2 and IFN-71. IL-10, IL-12 and IFN- are critical regulatory elements of cell-mediated immunity. IL-10 inhibits cellular immunity, while IL-12 and IFN- induce type-1 cytokine production and effective cell-mediated immunity72. PGE2 inhibits IFN-, IL-12 and induces high production of IL-4 and IL-1072. IL-10 overproduction at the tumor site is implicated in tumor-mediated immunosuppression73,74, enhanced angiogenesis75 and appears to be an PATEL & CHIPLUNKAR: COX-2 IN LUNG CANCER indicator of poor prognosis in NSCLC76. A correlation of increased serum IL-10 levels with clinical prognosis of NSCLC has been found76. COX-2 overexpression has been found in human lung cancer and tumor-derived high-level PGE2 production is reported to mediate dysregulation of host immunity by altering the balance of IL-10 and IL-12. Importantly, lung cancer-derived PGE2 induces a 10-100-fold increase in lymphocyte IL-10 production7. IL-10 augmentation by PGE2 in tumor bearing host mediates via phosphotyrosine kinase pathway7, but the specific down-stream molecular mechanism is not understood. Studies from our laboratory have shown involvement of p38 MAPK and ERK pathway in PGE2-mediated IL-10 production in NSCLC (Unpublished data). Specific inhibition of these pathways and COX-2 are accompanied by a significant decrement in IL-10 and a concomitant restoration of IFN- production, following TCR stimulation in these patients (Unpublished data). In normal cells, COX-2 expression is regulated by a negative feedback loop and elevated levels of IL-10 inhibit COX-2 expression, thereby limiting PGE2 production77. However, in NSCLC cells, COX-2 expression and subsequent PGE2 production is not down-regulated by IL-1078. This unresponsiveness of COX-2 to IL-10 may be due to the lack of surface expression of IL-10Rα, the IL-10 binding subunit78. In the lung tumor microenvironment, deficiency of an IL-10-mediated COX-2 regulatory feedback loop may contribute to COX-2 overexpression and maintenance of high levels of PGE2, thereby promoting the malignant phenotype. In contrast to NSCLC, normal lung epithelial cells display cell surface IL-10Rα, suggesting that NSCLC responsiveness to IL-10 may be regulated through modulation of cell surface IL-10Rα expression occurring during malignant transformation78. COX-2 in Regulation of Antigen Presentation DCs serve as professional APCs and are pivotal in the host immune response to tumor antigens. Antitumor immune responses require the coordinate activities of professional APCs and lymphocyte effectors. DCs acquire antigen in the periphery and subsequently transport it to lymphoid organs, where they prime specific immune responses64. The tumor microenvironment can adversely affect DC maturation and function74. Tumor-derived cytokines that mediate DC dysfunction include IL-10, IL-6, 425 vascular endothelial growth factor and macrophage colony stimulating factor74,79. COX-2 metabolites can play a major role in tumorinduced inhibition of DC differentiation80 and prostanoids mediate these effects by both IL-10dependent81 and independent pathways80. Tumorderived PGE2 has been shown to inhibit DC differentiation and function in an EP2 receptordependent manner82. Significant reduction in cell surface expression of CD11c, DEC205, MHC class I and II, CD80, CD86 on DCs cultured in the presence of tumor supernatant as well as a decrease in transporter associated proteins TAP1 and TAP2 have been reported by tumor secreted soluble mediators74. However these changes are not evident, when DCs are cultured in tumor supernatant from COX-2-inhibited tumors. Thus, COX-2 expression or activity of tumor induces suppression of DC activities and hence antigen presentation. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) Unlike corticosteroids, which inhibit phospholipase A2 activity, NSAIDs inhibit COX-1 and 2 activity or both and ultimately inhibit the formation of proinflammatory PGs. NSAIDs are widely used in pharmacology as anti-inflammatory, antipyretic, antithrombotic and analgesic agents. They can be selective COX-1 (SC-560, dexketoprofene, and mofezolac), COX-2 (celecoxib, rofecoxib, nimesulide, NS-398 and valdecoxib) and non-selective COX inhibitors (aspirin, indomethacin, diclofenac, piroxicam, diflunisal, and ibuprofen). All NSAIDs in clinical use inhibit COX reversibly by competing with the substrate AA for the active site of the enzyme, leading to a marked reduction in PG synthesis. However, aspirin causes irreversible acetylation of the cyclooxygenase component of COX, without affecting peroxidase activity83. The selective COX-2 inhibitors are preferred over non-selective COX inhibitors because of the ulcerogenic and renal side effects. The non-selective NSAIDs, such as aspirin and ibuprofen inhibit production of prostacyclin, which has a cytoprotective effect on the gastric mucosa and regulation of kidney function83. However, despite side effects are present, pharmacological properties of NSAIDs have been extensively exploited in numerous fields of clinical medicine since 19th century. In recent years, due to pivotal role of COX in development and growth of malignancies, interest has been aroused in a possible role for NSAIDs in prevention and treatment of 426 INDIAN J. BIOCHEM. BIOPHYS., VOL. 44, DECEMBER 2007 malignancies. Use of NSAIDs has shown to reduce the risk of colorectal, breast, cervical, esophageal, prostate and lung cancers48,84-87. Clinical Intervention of NSAIDs in NSCLC COX-2 inhibition has shown beneficial effect in attenuation of tumor development and activation of host immune responses in various in vitro and animal studies. Nimesulide, a COX-2 inhibitor can induce apoptosis in NSCLC cell lines. The IC50 values of various anticancer agents including etoposide and cisplatin are significantly reduced in vitro using nimesulide as an adjunct to chemotherapy in NSCLC48. This has also been validated in vivo. COX-2 inhibitors can significantly enhanced radiationinduced apoptosis in fibrosarcoma84 and such increase is also demonstrated in lung cancer models48,84,85. In NSCLC, COX-2 inhibitors potentiate radiation therapy in model systems by enhancing radiationinduced apoptosis86,87. Clinical trials are currently evaluating COX-2 inhibition in the context of treatment and chemoprevention of lung cancer. In clinical trial, with celecoxib a COX-2 inhibitor in combination with chemotherapy has shown significant decrease in PGE2 level within tumor tissues, suggesting that COX-2-dependent expression of genes, deleterious to the anti-tumor response, may also be decreased. Celecoxib combined with radiation therapy may also lead to changes in serum/plasma VEGF in gastric cancer88. The phase I and II trials are also in progress to evaluate the role of COX-2 inhibition at various stages of prostate, pancreatic, glioma and lung cancers86,87,89-92. Evidences, that COX-2 and EGFR have related signaling pathways that can interact to regulate cellular proliferation, migration and invasion in colon cancer and NSCLC44,93 has triggered interest in evaluating the combination of COX-2 and EGFR inhibition in NSCLC. There are various clinical trials in phase II or III in progress in NSCLC using COX-2 inhibitors. Risk against Benefit of NSAIDs The non-selective COX inhibitors are found to be associated with ulcerogenic and renal side effects, whereas COX-2 selective NSAIDs are associated with myocardial infarction and cardiac attack, but no ulcers. A leading hypothesis to explain the cardiovascular side effects related to COX-2 inhibition involve an imbalance between COX-2 derived prostacyclin (PGI2) and thromboxane A2 (TxA2). PGI2 and TxA2 both have opposing effects on platelets and smooth muscle cells in blood vessels. PGI2 inhibits platelet aggregation and causes vasodilation, conferring cardioprotective effects, whereas TxA2 induces platelet aggregation and causes vasoconstriction. Thus their balance is thought to be important for maintaining vascular homeostasis (i.e., preventing thrombosis and vasospasm, while performing efficient hemostasis). Inhibition of COX-2 derived PGI2 could result in a more thrombogenic state, leading to cardiovascular events. However, due to involvement of COX in various physiological and pathological conditions including various malignancies, there is considerable interest in the further potential clinical use of COX-2 inhibitors. The various clinical trials are going on with different NSAIDs alone or in combination to conventional treatment of cancer, in order to make tumors more sensitive to treatment and easy to eliminate. The phase II trials in NSCLC, prostate cancer, pancreatic and malignant glioma reveal that NSAIDs can be highly beneficial, when combined with radiotherapy or chemotherapy, with no or minimal cardiovascular side effects. In phase II trial, it has been demonstrated that celecoxib in combination with CPT-11 can be safely administered concurrently at full dose levels in patients with malignant glioma without any adverse effects91. Also, other phase II trials with celecoxib in combination with gemcitabine and radiation therapy in advanced pancreatic and prostate cancer respectively have shown low toxicity and good clinical response90,92. The future clinical trials would focus on establishing the relatively low risk therapeutic dose or combinations of NSAIDs that may be used in lung cancer or other malignancies. Another approach to minimize side effects is shifting the therapeutic target selection down-stream in the PG biosynthetic/response pathway towards specific PG synthase and receptors. These novel potential down-stream targets could be receptors for PGE2, PPAR, Wnt signaling molecules, microsomal prostaglandin E synthase (mPGES) and 15-hydroxyprostaglandin dehydrogenase (15-PDGH). Conclusion COX-2 is implicated in NSCLC in each step of multi-factorial process of tumor development, such as tumor progression, angiogenesis, metastasis and immune dysfunction. It has also shown an important role in regulation of cytokine balance, functions of PATEL & CHIPLUNKAR: COX-2 IN LUNG CANCER APCs and immune activation. Pre-clinical and clinical studies have demonstrated the benefits of NSAIDs in prevention and treatment of NSCLC. Early human trials are in progress to investigate COX-2 inhibition in chemoprevention and in combination with standard chemotherapy, radiation therapy and new biologic agents in regulation of NSCLC. Thus, involvement of COX in diverse regulatory mechanisms gives potential to COX inhibitors as therapeutic tools in management of lung cancer. References 1 Hida T, Yatabe Y, Achiwa H, Muramatsu H, Kozaki K, Nakamura S, Ogawa M, Mitsudomi T, Sugiura T & Takahashi T (1998) Cancer Res 58, 3761-3764 2 Achiwa H, Yatabe Y, Hida T, Kuroishi T, Kozaki K, Nakamura S, Ogawa M, Sugiura T, Mitsudomi T & Takahashi T (1999) Clin Cancer Res 5, 1001-1005 3 Hosomi Y, Yokose T, Hirose Y, Nakajima R, Nagai K, Nishiwaki Y & Ochiai A (2000) Lung Cancer 30, 73-81 4 Tsujii M & DuBois R N (1995) Cell 83, 493-501 5 Hsu A L, Ching T T, Wang D S, Song X, Rangnekar V M & Chen C S (2000) J Biol Chem 275, 11397-11403 6 Rao R, Redha R, Macias-Perez I, Su Y, Hwang M T, Hao C, Zent R, Breyer M D & Pozzi A (2007) J Biol Chem 7 Huang M, Stolina M, Sharma S, Mao J T, Zhu L, Miller P W, Wollman J, Herschman H & Dubinett S M (1998) Cancer Res 58, 1208-1216 8 Stolina M, Sharma S, Lin Y, Dohadwala M, Gardner B, Luo J, Zhu L, Kronenberg M, Miller P W, Portanova J, Lee J C & Dubinett S M (2000) J Immunol 164, 361-370 9 Dohadwala M, Batra R K, Luo J, Lin Y, Krysan K, Pold M, Sharma S & Dubinett S M (2002) J Biol Chem 277, 50828-50833 10 Niki T, Kohno T, Iba S, Moriya Y, Takahashi Y, Saito M, Maeshima A, Yamada T, Matsuno Y, Fukayama M, Yokota J & Hirohashi S (2002) Am J Pathol 160, 1129-1141 11 Su J L, Shih J Y, Yen M L, Jeng Y M, Chang C C, Hsieh C Y, Wei L H, Yang P C & Kuo M L (2004) Cancer Res 64, 554-564 12 Fitzpatrick F A & Soberman R (2001) J Clin Invest 107, 1347-1351 13 Samuelsson B & Hammarstrom S (1982) Vitam Horm 39, 1-30 14 Feng L, Sun W, Xia Y, Tang W W, Chanmugam P, Soyoola E, Wilson C B & Hwang D (1993) Arch Biochem Biophys 307, 361-368 15 Kurumbail R G, Stevens A M, Gierse J K, McDonald J J, Stegeman R A, Pak J Y, Gildehaus D, Miyashiro J M, Penning T D, Seibert K, Isakson P C & Stallings W C (1996) Nature 384, 644-648 16 Croxtal J D, Newman S P, Choudhury Q & Flower R J (1996) Biochem Biophys Res Commun 220, 491-495 17 Khuri F R, Wu H, Lee J J, Kemp B L, Lotan R, Lippman S M, Feng L, Hong W K & Xu X C (2001) Clin Cancer Res 7, 861-867 18 West K A, Castillo S S & Dennis P A (2002) Drug Resist Updat 5, 234-248 427 19 Kim H S, Youm H R, Lee J S, Min K W, Chung J H & Park C S (2003) Lung Cancer 42, 163-170 20 Park J M, Choi J E, Chae M H, Lee W K, Cha S I, Son J W, Kim C H, Kam S, Kang Y M, Jung T H & Park J Y (2006) BMC Cancer 6, 70 21 Wiese F W, Thompson P A & Kadlubar F F (2001) Carcinogenesis 22, 5-10 22 Ho I C & Lee T C (2002) J Toxicol Environ Hlth A 65, 245-263 23 Kelley D J, Mestre J R, Subbaramaiah K, Sacks P G, Schantz S P, Tanabe T, Inoue H, Ramonetti J T & Dannenberg A J (1997) Carcinogenesis 18, 795-799 24 Smith W L, DeWitt D L & Garavito R M (2000) Annu Rev Biochem 69, 145-182 25 Subbaramaiah K, Altorki N, Chung W J, Mestre J R, Sampat A & Dannenberg A J (1999) J Biol Chem 274, 10911-10915 26 Bissonnette M, Khare S, von Lintig F C, Wali R K, Nguyen L, Zhang Y, Hart J, Skarosi S, Varki N, Boss G R & Brasitus T A (2000) Cancer Res 60, 4602-4609 27 Wardlaw S A, March T H & Belinsky S A (2000) Carcinogenesis 21, 1371-1377 28 Williams C S, Tsujii M, Reese J, Dey S K & DuBois R N (2000) J Clin Invest 105, 1589-1594 29 Subbaramaiah K, Hart J C, Norton L & Dannenberg A J (2000) J Biol Chem 275, 14838-14845 30 Folkman J (2001) Semin Oncol 28, 536-542 31 Hanahan D & Folkman J (1996) Cell 86, 353-364 32 Ohta Y, Shridhar V, Bright R K, Kalemkerian G P, Du W, Carbone M, Watanabe Y & Pass H I (1999) Br J Cancer 81, 54-61 33 Gupta R A & Dubois R N (2001) Nat Rev Cancer 1, 11-21 34 Donnini S, Finetti F, Solito R, Terzuoli E, Sacchetti A, Morbidelli L, Patrignani P & Ziche M (2007) Faseb J 35 Shao J, Jung C, Liu C & Sheng H (2005) J Biol Chem 280, 26565-26572 36 Ballou L R (2002) Adv Exp Med Biol 507, 585-591 37 Okada F, Rak J W, Croix B S, Lieubeau B, Kaya M, Roncari L, Shirasawa S, Sasazuki T & Kerbel R S (1998) Proc Natl Acad Sci (USA) 95, 3609-3614 38 Marrogi A J, Travis W D, Welsh J A, Khan M A, Rahim H, Tazelaar H, Pairolero P, Trastek V, Jett J, Caporaso N E, Liotta L A & Harris C C (2000) Clin Cancer Res 6, 4739-4744 39 Pold M, Zhu L X, Sharma S, Burdick M D, Lin Y, Lee P P, Pold A, Luo J, Krysan K, Dohadwala M, Mao J T, Batra R K, Strieter R M & Dubinett S M (2004) Cancer Res 64, 1853-1860 40 Tsujii M, Kawano S & DuBois R N (1997) Proc Natl Acad Sci (USA) 94, 3336-3340 41 Dohadwala M, Luo J, Zhu L, Lin Y, Dougherty G J, Sharma S, Huang M, Pold M, Batra R K & Dubinett S M (2001) J Biol Chem 276, 20809-20812 42 Yu Q & Stamenkovic I (2000) Genes Dev 14, 163-176 43 Shankavaram U T, Lai W C, Netzel-Arnett S, Mangan P R, Ardans J A, Caterina N, Stetler-Stevenson W G, BirkedalHansen H & Wahl L M (2001) J Biol Chem 276, 19027-19032 44 Coffey R J, Hawkey C J, Damstrup L, Graves-Deal R, Daniel V C, Dempsey P J, Chinery R, Kirkland S C, DuBois R N, Jetton T L & Morrow J D (1997) Proc Natl Acad Sci (USA) 94, 657-662 428 INDIAN J. BIOCHEM. BIOPHYS., VOL. 44, DECEMBER 2007 45 Hoang B, Zhu L, Shi Y, Frost P, Yan H, Sharma S, Sharma S, Goodglick L, Dubinett S & Lichtenstein A (2006) Blood 107, 4484-4490 46 Stacker S A, Caesar C, Baldwin M E, Thornton G E, Williams R A, Prevo R, Jackson D G, Nishikawa S, Kubo H & Achen M G (2001) Nat Med 7, 186-191 47 Barnes N L, Warnberg F, Farnie G, White D, Jiang W, Anderson E & Bundred N J (2007) Br J Cancer 96, 575-582 48 Hida T, Kozaki K, Muramatsu H, Masuda A, Shimizu S, Mitsudomi T, Sugiura T, Ogawa M & Takahashi T (2000) Clin Cancer Res 6, 2006-2011 49 Van Putten V, Refaat Z, Dessev C, Blaine S, Wick M, Butterfield L, Han S Y, Heasley L E & Nemenoff R A (2001) J Biol Chem 276, 1226-1232 50 Liu X H, Yao S, Kirschenbaum A & Levine A C (1998) Cancer Res 58, 4245-4249 51 Hara A, Yoshimi N, Niwa M, Ino N & Mori H (1997) Jpn J Cancer Res 88, 600-604 52 Lim E S, Rhee Y H, Park M K, Shim B S, Ahn K S, Kang H, Yoo H S & Kim S H (2007) Ann N Y Acad Sci 1095, 7-18 53 Baratelli F, Krysan K, Heuze-Vourc'h N, Zhu L, Escuadro B, Sharma S, Reckamp K, Dohadwala M & Dubinett S M (2005) J Leukoc Biol 78, 555-564 54 Burnet F M (1970) Prog Exp Tumor Res 13, 1-27 55 Balkwill F & Mantovani A (2001) Lancet 357, 539-545 56 Vane J R, Bakhle Y S & Botting R M (1998) Annu Rev Pharmacol Toxicol 38, 97-120 57 Tilley S L, Coffman T M & Koller B H (2001) J Clin Invest 108, 15-23 58 Rocca B, Spain L M, Ciabattoni G, Patrono C & FitzGerald G A (1999) J Immunol 162, 4589-4597 59 Rocca B, Spain L M, Pure E, Langenbach R, Patrono C & FitzGerald G A (1999) J Clin Invest 103, 1469-1477 60 Rossi Paccani S, Boncristiano M & Baldari C T (2003) Cell Mol Life Sci 60, 1071-1083 61 Ullman K S, Northrop J P, Verweij C L & Crabtree G R (1990) Annu Rev Immunol 8, 421-452 62 Iniguez M A, Punzon C & Fresno M (1999) J Immunol 163, 111-119 63 Paccani S R, Boncristiano M, Ulivieri C, D'Elios M M, Del Prete G & Baldari C T (2002) J Biol Chem 277, 1509-1513 64 Banchereau J & Steinman R M (1998) Nature 392, 245-252 65 Norgauer J, Ibig Y, Gmeiner D, Herouy Y & Fiebich B L (2003) Int J Mol Med 12, 83-86 66 Gosset P, Bureau F, Angeli V, Pichavant M, Faveeuw C, Tonnel A B & Trottein F (2003) J Immunol 170, 4943-4952 67 Coussens L M & Werb Z (2002) Nature 420, 860-867 68 Alleva D G, Burger C J & Elgert K D (1994) J Immunol 153, 1674-1686 69 Wang T, Sheppard J R & Foker J E (1978) Science 201, 155-157 70 Benbernou N, Esnault S, Shin H C, Fekkar H & Guenounou M (1997) Immunology 91, 361-368 71 Munoz E, Zubiaga A M, Merrow M, Sauter N P & Huber B T (1990) J Exp Med 172, 95-103 72 Wu J, Cunha F Q, Liew F Y & Weiser W Y (1993) J Immunol 151, 4325-4332 73 Hagenbaugh A, Sharma S, Dubinett S M, Wei S H, Aranda R, Cheroutre H, Fowell D J, Binder S, Tsao B, Locksley R M, Moore K W & Kronenberg M (1997) J Exp Med 185, 2101-2110 74 Sharma S, Stolina M, Lin Y, Gardner B, Miller P W, Kronenberg M & Dubinett S M (1999) J Immunol 163, 5020-5028 75 Hatanaka H, Abe Y, Naruke M, Tokunaga T, Oshika Y, Kawakami T, Osada H, Nagata J, Kamochi J, Tsuchida T, Kijima H, Yamazaki H, Inoue H, Ueyama Y & Nakamura M (2001) Clin Cancer Res 7, 1287-1292 76 Hatanaka H, Abe Y, Kamiya T, Morino F, Nagata J, Tokunaga T, Oshika Y, Suemizu H, Kijima H, Tsuchida T, Yamazaki H, Inoue H, Nakamura M & Ueyama Y (2000) Ann Oncol 11, 815-819 77 Pomini F, Caruso A & Challis J R (1999) J Clin Endocrinol Metab 84, 4645-4651 78 Heuze-Vourc'h N, Zhu L, Krysan K, Batra R K, Sharma S & Dubinett S M (2003) Cancer Res 63, 766-770 79 Caux C (1998) Eur J Dermatol 8, 375-384 80 Sombroek C C, Stam A G, Masterson A J, Lougheed S M, Schakel M J, Meijer C J, Pinedo H M, van den Eertwegh A J, Scheper R J & de Gruijl T D (2002) J Immunol 168, 4333-4343 81 Harizi H, Juzan M, Pitard V, Moreau J F & Gualde N (2002) J Immunol 168, 2255-2263 82 Yang L, Yamagata N, Yadav R, Brandon S, Courtney R L, Morrow J D, Shyr Y, Boothby M, Joyce S, Carbone D P & Breyer R M (2003) J Clin Invest 111, 727-735 83 van der Ouderaa F J, Buytenhek M & van Dorp D A (1980) Adv Prostaglandin Thromboxane Res 6, 139-144 84 Milas L, Hunter N, Furuta Y, Nishiguchi I & Runkel S (1991) Int J Radiat Biol 60, 65-70 85 Milas L, Ito H, Nakayama T & Hunter N (1991) Cancer Res 51, 3639-3642 86 Choy H & Milas L (2003) J Natl Cancer Inst 95, 1440-1452 87 Komaki R, Liao Z & Milas L (2004) Semin Oncol 31, 47-53 88 Zhou Y, Ran J, Tang C, Wu J, Honghua L, Xingwen L, Ning C & Qiao L (2007) Cancer Biol Ther 6, 269-275 89 Reckamp K L, Krysan K, Morrow J D, Milne G L, Newman R A, Tucker C, Elashoff R M, Dubinett S M & Figlin R A (2006) Clin Cancer Res 12, 3381-3388 90 Ferrari V, Valcamonico F, Amoroso V, Simoncini E, Vassalli L, Marpicati P, Rangoni G, Grisanti S, Tiberio G A, Nodari F, Strina C & Marini G (2006) Cancer Chemother Pharmacol 57, 185-190 91 Reardon D A, Quinn J A, Vredenburgh J, Rich J N, Gururangan S, Badruddoja M, Herndon J E, 2nd, Dowell J M, Friedman A H & Friedman H S (2005) Cancer 103, 329-338 92 Pruthi R S, Derksen J E, Moore D, Carson C C, Grigson G, Watkins C & Wallen E (2006) Clin Cancer Res 12, 2172-2177 93 Buchanan F G, Wang D, Bargiacchi F & DuBois R N (2003) J Biol Chem 278, 35451-35457