Appendix S1: Laboratory procedure to sequence a fragment of the
advertisement

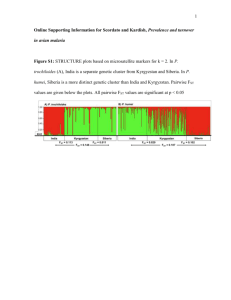
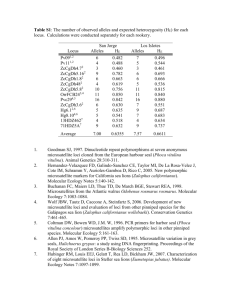
Appendix S1: Laboratory procedure to sequence a fragment of the mtDNA control region We used primers MelCR1 and MelCR6 (Marmi et al. 2006) to amplify a 594-base-pair fragment of the 5’-end of the control region of 327 individuals (Table S1). We amplified fragments using 10-μl polymerase chain reactions (PCRs) that contained approximately 10 ng of DNA and 0.5 units of Biotaq DNA Polymerase (Bioline, London, UK) in the manufacturer’s buffer with a concentration of 1.0 μM of each primer, 1.5 mM MgCl2 and 0.1 mM of each dNTP. The PCRs were performed in a thermal cycler (MJ Research, St. Bruno, Canada) using the following procedure: one cycle of 3 min at 95°C, followed by 35 cycles of 94°C for 30s, 57°C for 45 s, 72°C for 30s, and a final extension at 72°C for 10 min. Successful PCR products were precipitated with ethanol and forward and reverse strands sequenced using Big Dye Terminator chemistry (Applied Biosystems, Carlsbad, CA, USA) on an ABI 3730 capillary DNA automated sequencer (Applied Biosystems). Appendix S2: Information on the composition and reaction times of the microsatellite multiplex reactions. The composition of the Multiplex 1 contained loci Mel101, Mel103, Mel104, Mel106, Mel107, Mel108 (Carpenter et al. 2003) and Mel10 (Dominga-Roura et al. 2003). Multiplex 2 contained loci Mel102, Mel112, Mel113, Mel114, Mel115 and Mel117 (Carpenter et al. 2003). Multiplex 3 contained loci Mel110 (Carpenter et al. 2003), Mel1, Mel10, Mel12, Mel14 and Mel15 (DomingaRoura et al. 2003). We amplified each multiplex using the Qiagen multiplex Kit (Qiagen, Hilden, Germany). Each multiplex reaction contained 1 x Qiagen multiplex master mix, 0.2 μM of each primer and 0.5 x Q-solution. After drying 1 μl of DNA (c. 10ng/ml) for 15 min at 52 °C in a 384-well PCR plate (Greiner Bio-One, Stonehouse, UK), multiplex reactions were performed in a total volume of 2 ml. Following reaction times described in the multiplex kit manual, a touch-down profile was used, starting with a 15-min denaturation at 95 °C, followed by denaturation at 94 °C for 30 s, annealing at initially 61 °C for 90 s and extension at 72 °C for 1min. The annealing temperature was reduced by 1 °C per cycle for five cycles then kept at 55 °C for the remaining 29 cycles. Final incubation was at 60 °C for 30 min. Reactions were performed using a DNA Engine Tetrad thermocycler (MJ Research, Waltham, MA, USA). PCR products were separated using an ABI 3730 automated DNA sequencer (Applied Biosystems, Warrington, UK) and the data were analysed using GENEMAPPER version 3.7 (Applied Biosystems). We followed a multiple tubes approach (Frantz et al. 2003) to genotype the hair samples. The genetic profiles of all samples consisted of at least 12 loci. Appendix S3: Parameters of the Bayesian genetic clustering analyses First, we analysed the data using the program STRUCTURE v. 2.3.1 (Pritchard et al. 2000). To estimate the number of subpopulations (K), 10 independent runs of K = 1–15 were carried out with 106 Markov chain Monte Carlo (MCMC) iterations after a burn-in period of 105 iterations, using the model with correlated allele frequencies and assuming admixture. ALPHA, the Dirichlet parameter for the degree of admixture, was allowed to vary between runs. We decided on the most probable number of sub-populations based on the log-likelihood values (and their convergence) associated with each K. For each individual, the probability of population membership was manually averaged across the 10 runs of the optimal K. Second, we analysed the population genetic structure using GENELAND v.3.2.4 (Guillot et al. 2005). The number of genetic clusters was determined by running the algorithm ten times, allowing K to vary, with the following parameters: 106 MCMC iterations with a thinning of 1000, maximum rate of the Poisson process fixed to 100, minimum K = 1, maximum K = 15 and maximum number of nuclei in the Poisson–Voronoi tessellation fixed to 300. The Dirichlet model was used as a prior for all allele frequencies. After inferring the number of populations, the algorithm was run a further 100 times with K fixed to the inferred number of clusters (K=9), with 2.5x105 MCMC iterations with a thinning of 250 and with the other parameters the same as above. Excluding the first 100 values as a burn-in, the mean logarithm of the posterior probability was calculated for each of the 100 runs, and the posterior probability of population membership for each pixel of the spatial domain was then computed for the ten runs with the highest values. For each individual, the probability of population membership was manually averaged across these 10 runs. Appendix references Carpenter PJ, Dawson DA, Greig C, Parham A, Cheeseman C, Burke, T. (2003) Isolation of 39 polymorphic microsatellite loci and the development of a fluorescently labelled marker set for the Eurasian badger (Meles meles) (Carnivora: Mustelidae). Molecular Ecology Notes, 3, 610-615. Domingo-Roura X, Macdonald DW, Roy MS, Marmi J, Terradas J, Woodroffe R, Burke T, Wayne RK (2003) Confirmation of low genetic diversity and multiple breeding females in a social group of Eurasian badgers from microsatellite and field data. Molecular Ecology, 12, 533–539. Frantz AC, Pope LC, Carpenter PJ, Roper TJ, Wilson GJ, Delahay RJ, Burke T. (2003) Reliable microsatellite genotyping of the Eurasian badger (Meles meles) using faecal DNA. Molecular Ecology, 12, 1649–1661. Guillot G, Estoup A, Mortier F, Cosson J (2005) A spatial statistical model for landscape genetics. Genetics, 170, 1261-1280. Marmi J, Lopez-Giraldez F, Macdonald D et al. (2006) Mitochondrial DNA reveals a strong phylogeographic structure in the badger across Eurasia. Molecular Ecology, 15, 1007-1020. Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics, 155, 945–959.