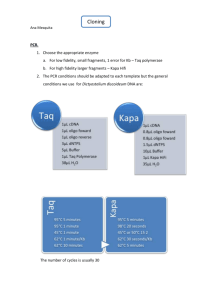
PCR Mutagenesis:
advertisement

PCR Mutagenesis Mara Duncan (3/99) modified from M.Stark (Yeast Gene Analysis: Ed. Brown and Tuite.) This protocol relies on the ability of yeast to repair a gapped plasmid with a second homologous piece of DNA. This protocol requires that you can gap a plasmid containing the DNA to be mutagenized within the region to be mutagenized. This gap does not have to span the entire region to be mutagenized. After identifying restriction enzymes that will produce such a gap, choose primers that define the entire region to be mutagenized and are 50-100 bp outside the gap. For a better description, read the original protocol. One trick is to PCR off of a plasmid that cannot replicate in yeast so you don't have to gel purify the PCR fragment. 1. PCR with the following conditions: 0.2mM DNTP's 4ng/µl primers 0.1-0.2ng/ul template 2mM MgCl2 0.05 U/µl Taq Do ten separate PCR reactions. Do at least 30 cycles. There are other conditions that you can vary to raise % mutagenesis such as adding MnCl2 or skewing DNTP concentrations, but high levels of Taq for several cycles provides some mutagenesis and enough PCR product to get good transformation efficiency. 2. With a Quiagen PCR-Purification Kit, clean up the each reaction separately. 3. Digest gapped plasmid with appropriate enzyme and gel purify. 3. Quantitate DNA from PCR and digest on a gel. 4. Co-transform 500ng PCR products and 100ng vector into yeast. Do one transformation reaction per PCR reaction. 5. Resuspend transformation in 1ml selective media. 6. Plate 100µl on selective plates (ie Leu- if mutagenized plasmid is LEU+). Save the rest of transformation at 4°C. Grow plate at permissive temperature 3-5 days. 7. Once the first plate has grown up determine correct dilution/concentration for the rest of the transformation to give ~200 colonies/plate.