Supporting Information_revised
advertisement

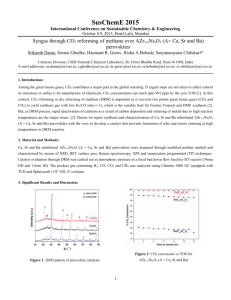
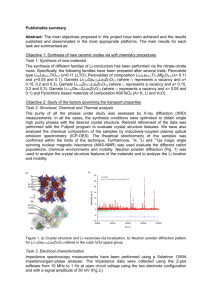
Supporting Information Hole-Transport Material Variation in Fully Vacuum Deposited Perovskite Solar Cells Lauren E. Polander, Paul Pahner, Martin Schwarze, Matthias Saalfrank, Christian Koerner, Karl Leo* Institut für Angewandte Photophysik, Technische Universität Dresden, 01069 Dresden, Germany Contents Experimental Details Materials Page S2 General Device Fabrication Page S2 Perovskite Layer Deposition Page S3 Profilometry Page S3 Grazing Incidence X-Ray Diffraction Page S4 Ultraviolet Photoelectron Spectroscopy Page S5 Conductivity Measurements Page S5 Device Characteristics Page S6 References 1 Page S2 Page S6 Experimental Details Materials. All reagents were purchased from Aldrich and used as received unless otherwise stated. Methylammonium iodide (CH3NH3I) was prepared from methylamine and hydroiodic acid as reported1 and purified by recrystallization in ethanol and isopropanol. Buckminster fullerene (C60) was purchased from Bucky, USA, and purified twice by thermal gradient sublimation. 2,2',7,7'Tetrakis[N,N-di(4-methoxyphenyl)amino]-9,9'-spirobifluorene (Spiro-MeO-TAD, 1) was purchased from Feiming Chemical Limited and used as received. N4,N4,N4',N4'-Tetrakis(4-methoxyphenyl)[1,1'-biphenyl]-4,4'-diamine spirobifluorene (MeO-TPD, (Spiro-MeO-TPD, 3), 2), 2,7-bis[N,N-bis(4-methoxy-phenyl)amino]-9,9- 2,2',7,7'-tetrakis-[N,N-di(4-methylphenyl)amino]-9,9'- spirobifluorene (Spiro-TTB, 4), 2,2',7,7'-tetrakis-(N,N-diphenylamino)-9,9'-spirobifluorene (SpiroTAD, 5), and 9,9-bis[4-(N,N-bis-biphenyl-4-yl-amino)phenyl]-9H-fluorene (BPAPF, 6) were purchased from Lumtec and purified twice by gradient sublimation. 2,2'-(Perfluoronaphthalene-2,6diylidene)dimalononitrile (F6-TCNNQ) and NDP9 were purchased from Novaled GmbH and used as received. General Device Fabrication. Thin film samples were prepared by thermal vapor deposition in ultrahigh vacuum (UHV) with a base pressure at or below 10–7 mbar. The layer thicknesses were determined during evaporation using quartz crystal monitors calibrated for the respective material prior to evaporation. The glass/ITO substrates (Corning Eagle XG, 1.1 mm, Thin film devices, USA) were carefully cleaned with NMP, ethanol, and oxygen plasma before being transferred into the vacuum chamber. One general stack design was used for all cells as depicted in Figure S1. Layers 1–3 were deposited by thermal evaporation in a UHV system (K.J. Lesker, U.K.). The substrates were transported in a nitrogen-sealed transfer box to a second UHV chamber for the deposition of layer 4 (CH3NH3PbI3–xClx perovskite deposition detailed below) and again, via nitrogen-filled glovebox, to a third UHV chamber for the deposition of layers 5 and 6 (CreaPhys GmbH, Germany). The completed solar cells were encapsulated with a transparent encapsulation glass, fixed by UV-hardened epoxy glue, in a nitrogen-filled glovebox. Figure S1. General device architecture for all cells in the study. Perovskite in layer 4 refers to the methylammonium lead iodide/chloride perovskite of the form CH3NH3PbI3–xClx. 2 Perovskite Layer Deposition. The perovskite layer deposition was performed in a custom-made vacuum system (CreaPhys GmbH, Germany) by thermal co-evaporation of PbCl2 and CH3NH3I at a base pressure of 10−7 mbar. The deposition ratio and layer thickness were monitored using calibrated quartz crystal microbalances (QCM, Thickness/Rate Monitor STM-100/MF, Syncon Instrument). Three QCM sensors are present in the chamber, one monitoring the rate of each evaporation source and a third at the height of the substrate holder. The PbCl2 source QCM was used to calibrate the deposition rate of PbCl2 using normal tooling procedures (ρ = 5.85 mg cm–3). However, a fluctuating signal was observed at each of the three QCMs during heating of the CH3NH3I source and the deposition rate of this material could not be determined using normal methods. Alternatively, the amount of CH3NH3I in the chamber was monitored by stabilizing the chamber pressure at ca. 1.0×10–4 mbar. The third QCM placed at substrate level was used to monitor the perovskite layer formation. Initially, this QCM was calibrated by tooling with PbCl2. However, as the density and exact composition of the final film is unknown, the thickness of this layer was more accurately determined using profilometry measurements (Figure S2, details below). After loading the sources, the chamber was evacuated and allowed to stabilize overnight. To begin evaporation, the CH3NH3I crucible was heated slowly until a pressure of ca. 1.0×10–4 mbar could be maintained in the chamber (crucible temperature of 130–160 °C). The PbCl2 source was then heated to achieve a rate of ca. 0.5–0.7 Å s−1 (360–380 °C) at the PbCl2 QCM, which corresponded to a deposition rate at the substrate QCM of ca. 0.8–1.0 Å s−1. The films were dark brown when removed from the vacuum chamber and no annealing treatments were performed. Formation of a perovskite film was confirmed using grazing incidence Xray diffraction (GIXRD) for a sample deposited on glass (Figure S3, details below). Figure S2. Profilometer trace of a vacuum deposited perovskite layer on glass with silver overlay. Profilometry. The perovskite layer thickness calibration was carried out by evaluating the actual thickness of deposited layers using a profilometer (Dektak 150, Veeco, CA). A mask was used to prepare a sample with a sharp perovskite layer edge on a glass substrate. Subsequently a thin silver layer was evaporated on top (without mask) to enable scanning of a more rigid surface and avoid 3 direct air exposure of the perovskite. A constant force of 3.0 mg was applied to the stylus (5.0 µm radius) while measuring the height profile of the step-like surface. Several scans were performed at different areas on the sample (Figure S2) and it was determined that the average roughness was approximately 6 nm and the film thickness was ca. 195 nm (corresponding to a reading of 100 nm at the substrate QCM monitor). This correction factor was used for the thickness calibration throughout the study. Grazing Incidence X-Ray Diffraction (GIXRD). The thin-film perovskite sample was measured using a Bruker D8 Discover diffractometer in GIXRD geometry. For the thin-film measurements, Cu Kα radiation from an X-ray tube operated at 40kV and 40mA was paralleled by a 3rd generation 60mm Göbel mirror. The diffracted beam was detected by a scintillation counter after passing a parallel plate collimator (0.35° angular acceptance). The diffraction pattern of the film was collected by variation of the 2θ detector angle in the range of 3–90° with an angle step width Δ2θ = 0.1° and 30s sampling time. The measurements were carried out at two different incidence angles in order to eliminate air scattering contributions. The first measurement was taken at Ω = 0.3°, slightly above the critical angle of total external reflection. At this incidence angle, the penetration depth is in the range of the perovskite film thickness and therefore contributions from the glass substrate are assumed to be negligible. The second measurement was taken at Ω = 0.07°, well below the critical angle, and was used as background correction for the first measurement. Figure S3. Grazing incidence X-ray diffraction of a vacuum deposited perovskite layer on glass. Diffraction peaks at 14.2°, 28.6°, and 43.3° are assigned to the (110), (220), and (330) planes of a tetragonal perovskite structure, respectively. The typical tetragonal structure for CH3NH3MX3 hybrid perovskites (M=Pb, Sn; X=Cl, Br,I) consists of a three-dimensional anionic framework of PbX6 (X = I, Cl) octahedra with methylammonium cations in the interstitial space.2 Additional peaks are present in the diffractogram as shown in Figure S3. These are believed to correspond to imperfections in the perovskite deposition, and particularly to excess PbX2 present in the sample. Although the evaporation conditions were held constant, slight fluctuations could be seen in the relative PbX 2 4 content from sample-to-sample, which presumably also effected the solar cell performance. For this reason, multiple substrates were fabricated and evaluated for each HTM in the study. Ultraviolet Photoelectron Spectroscopy (UPS). The organic layers (ca. 10–12 nm) were thermally evaporated at a pressure of 10–8 mbar on sputter cleaned metal foils or plasma cleaned ITO and transferred in UHV to the UPS chamber. UPS spectra were acquired from He I (21.22 eV) excitation lines using a PHOIBOS 100 system (Specs, Berlin, Germany) at base pressure of 10 –11 mbar. All experiments were calibrated to the Fermi edge of an atomically clean gold surface. UPS traces for HTMs 2–6 are shown in Figure S4 and an energy level diagram for the full system is shown in Figure 1 of the main text. The values for the perovskite layer and HTM 1 are as reported.3 Figure S4. UPS measurements on HTM 2 (—), 3 (—), 4 (—), 5 (—), and 6 (—). The spectra are calibrated to vacuum level position and scaled to the same intensity. The IP values are defined as the onset of the highest occupied molecular orbital as shown in the graph. Conductivity Measurements. The conductivity measurements of the HTMs were performed in situ during quartz-crystal controlled co-evaporation of matrix and dopant molecules at a pressure ca. 10–8 mbar. The current increase was measured between the contacts of a meander-like structure (1 nm Cr / 40 nm Au) upon increasing layer thickness of the matrix:dopant films in the channel (b = 500 µm, l = 0.111 m) during deposition. The sign of the applied voltage (10 V) did not alter the magnitude of the current (Ohmic behavior). Due to a broad distribution of conductivities among the chosen HTMs, doping techniques were used to establish matrix layers with comparable conductivities. Each material was initially tested at 2 wt% doping of either F6-TCNNQ (HTMs 1–5) or NDP9 (HTM 6). In the case of HTM 5 and 6, this doping 5 ratio resulted in conductivities well below the range of the other HTMs (σ < 1×10–5 S cm–1). Therefore, an increased doping ratio of 10 wt% was used for these materials in order to achieve comparable conductivity values (Figure S5). Figure S5. Comparison of the conductivity values for HTMs 1–6. The black bars indicate the doping ratios used for device fabrication. The weight percent and molar ratio of dopant (F 6-TCNNQ) in the matrix material are noted for HTM 1–5. For HTM 6, only the weight percent is noted due to the use of a proprietary dopant (NDP9). Device Characterization. The J–V characteristics were recorded with a Keithley 2400 source measure unit using a sun simulator 16S-003-300 (Solarlight Company Inc.) that was tuned to mismatch corrected light intensity, determined by an outdoor reference cell (Fraunhofer Insitute for Solar Energy Systems, Freiburg, Germany). Mismatch correction was performed using the measured EQE spectrum, the EQE of the reference diode, and the illumination spectrum of the sun simulator, which is recorded with every J–V measurement.4 The active area of the solar cells was determined using optical microscopy to be approximately 6.44 mm2. For the EQE measurement, a homemade setup based on a zenon arc lamp, a monochromator (Cornerstone 260), and a lock-in amplifier (Signal Recovery SR 7265) was used. The setup was calibrated with a reference diode (Hamamatsu S1337). References (1) Lee, M. M.; Teuscher, J.; Miyasaka, T.; Murakami, T. N.; Snaith, H. J. Science 2012, 338, 643. (2) Mitzi, D. B. Prog. Inorg. Chem. 1999, 48, 1. (3) Schulz, P.; Edri, E.; Kirmayer, S.; Hodes, G.; Cahen, D.; Kahn, A. Energy Environ. Sci. 2014, 7, 1377. (4) Shrotriya, V.; Li, G.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. Adv. Funct. Mater. 2006, 16, 2016. 6