snphap
advertisement

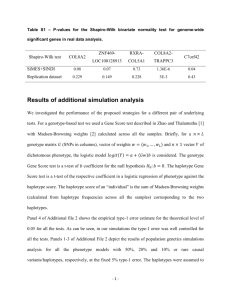
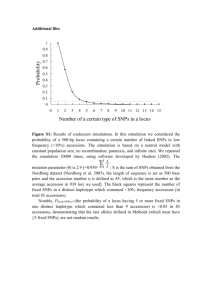
- SNPHAP A program for estimating frequencies of large haplotypes of SNPs ================================================================ (Version 1.0) David Clayton Department of Medical Genetics Cambridge Institute for Medical Research Wellcome Trust/MRC Building Addenbrooke's Hospital, Cambridge, CB2 2XY Disclaimer ---------This program has now had a fair bit of use by our group and others. However it comes with no guarantees. I'd like to know of any difficulties/bugs that people experience and will try and fdeal with them, but this may take some time. Introduction -----------This program implements a fairly standard method for estimating haplotype frequencies using data from unrelated individuals. It uses an EM algorithm to calculate ML estimates of haplotype frequencies given genotype measurements which do not specify phase. The algorithm also allows for some genotype measurements to be missing (due, for example, to PCR failure). It also allows multiple imputation of individual haplotypes. EM algorithm -----------The well-known algorithm first expands the data for each subject into the complete set of (fully phased) pairs of haplotype consistent with the observed data (the "haplotype instances"). The EM algorithm then proceeds as follows: E step: Given current estimates of haplotype probabilities and assuming Hardy-Weinberg equilibrium, calculate the probability of each phased and complete genotype assignment for each subject. Scale these so that they sum to 1.0 within each subject. These are then the POSTERIOR PROBABILITIES of the genotype assignments. M step: Calculate the next set of estimates of haplotype probabilities by summing posterior probabilities over instances of each distinct haplotype. After scaling to sum to 1.0, These provide the new estimates of the PRIOR PROBABILITIES. This algorithm is widely used and not novel. However, for a large number of loci, the number of possible haplotype instances rapidly becomes impossibly large, even though the eventual solution may only give appreciable support to a rather limited set of haplotypes. This program avoids this difficulty by starting by fitting 2-locus haplotypes and extending the solution by one locus at a time. As each new locus is added, the number of haplotype instances to be considered is first expanded, by considering all possible larger haplotypes. Then, after applyiny the EM algorithm to estimate the "prior" haplotype probabilities and the posterior probilities of haplotype instances, the haplotype instances are culled in two ways: Posterior trimming: Any genotype assignment whose posterior probability falls below a given threshold is deleted and the posterior probabilities of assignments of genotype to the subject are recomputed. Prior trimming: All instances of any haplotype whose prior probability falls below a threshold are removed. This option is not used by default since it can lead to difficulties in comparing likelihoods (see below). We add one locus at a time until completion. The process of culling haplotype assignments at early stages can lead to solutions which are not optimal. For example, haplotype 1.1.1 may have zero estimated frequency in the maximum likelihood analysis of the three-locus haplotype, while 1.1.1.x may have non-zero estimated frequency in the ML solution to the four-locus problem. It is not clear how often this will be a problem. A partial solution is to try including loci in different orders, seeing if the soultion obtained varies. A further protection is not to cull haplotypes after inclusion of every locus, but only every k loci, although there will be a penalty both in computer time and use of memory incurred by choice of large values of k. Multiple imputation ------------------Sampling the (Bayesian) posterior distribution of individual haplotype data is conveniently carried out using a Gibbs sampler. This mimics the EM algorithmm but uses stochastic steps rather than deterministic ones. It has been termed the IP (Imputation/Posterior sampling) algorithm. In our case the algorithm works as follows: I-step (replaces the E-step): For each subject, pick a haplotype assignment from the possible instances, with probability given by the current posterior, or "full conditional" distribution. P-step (replaces the M-step): Sample the haplotype population frequencies from their full conditional Bayesian posterior. If the prior is a Dirichlet distribution with constant df on all possible haplotypes*, the full conditional posterior distribution is also Dirichlet. To obtain the set of df parameters for this posterior Dirichlet, we simply add the constant prior df to the number of chromosomes currently assigned to each haplotype. (* i.e. if the unknown population haplotype relative frequencies are denoted p_1, p2, ..., p_i, ..., p_n, then their prior density is assumed to be proportional to n Product i=1 d-1 (p_i) where d is the prior degree of freedom parameter) There can be difficulties in sampling the entire space using this algorithm if the prior Dirchlet df is taken as zero; if a haplotype is not assigned to any individual at in one step, then the full conditional posterior is improper and the haplotype will be given zero probability at the next step. Thereafter it can never be sampled again. Also, when there are multiple maxima in the likelihood, the algorithm may become "stuck" under one peak. To avoid these difficulties, provision is made to start the prior df parameter at a relatively large value, thereby giving all haplotypes an appreciable probability of being sampled. Thereafter the prior df parameter is reduced at each step. This algorithm is repeated for a fixed number of steps to obtain a single imputation. The prior df parameter is then set back up to the high value, the population haplotype frequencies restored to their MLE's, and the process repeated to obtain the next imputation. And so on. Warning: Although multiple imputation using the IP algorithm is an established technique (see Schafer J.L. "Analysis of Incomplete Multivariate Data" Chapman and Hall: London, 1997), it remains to be rigorously validated in this application. Multiple maxima --------------It is well known that the likelihood surface for this problem may have multiple maxima and that the EM algorithm will only converge to a local maximum. After all loci have been added and a final trimmed list of haplotype instances has been computed, the EM algorithm may be repeated multiple times from random starting points in order to search for the global maximum. The random starting points may be chosen in one of two ways: (a) from randomly chosen values for the prior haplotype probabilities, or (b) from randomly chosen posterior probabilities for each haplotype assignment. Random starting points can also be chosen in the first set of EM iterations and, in this case, method (b) is used. Use --The program is invoked from the command line by snphap [-ds # -de # -i # -l # -mb # -mc # -mi # -mm # -n -pr # -ro -rv -rp -rs -sd # -ss -to # -th #] input-file [output-file-1] [output-file-2] -po # The input file should contain the data in subject order, with a subject identifier followed by pairs of alleles of each locus. The subject identifier need not be numeric, but must not include "white space" (blanks or tabs). The alleles should either be coded 1, 2 (numeric coding), or A,C,G or T ("nucleotide" coding). Missing data is indicated by 0 in numeric coding and, for nucleotide coding, by any character not hitherto mentioned. Data fields should be separated by any "white space" (any number of blanks, tabs or new-line characters). By default loci, are added in the same order that they appear on the input file but, optionally, they may be added in reverse order or random order. The log likelihood output from this program should be used with some caution, particularly when prior trimming has been applied, since likelihoods which do not consider the same subsets of possible haplotypes may not be comparable. Options ------The optional flags allow one to set the following parameters: -i # The maximum number of EM iterations at each step is # (default 50). -l # The number of loci is #. This option is no longer necessary if the data are entered as one line per subject. -k # Kill improbable haplotype assignments (see -pr and -po) after every # loci (default is 5). -mb # The maximum amount of dynamic storage to be allocated to the program is # Mbytes. This option should not be needed since, by default, the program should be able to determine this. -mi # Create # output files containing fully phased genotypes imputed at random from the posterior distribution. -mm # Carry out the final EM maximization # times, starting from random starting points. The solution with the largest likelihood is accepted. -nu data Forces numeric coding (1/2) of alleles on output, even when input are in A/T/C/G format (default is "off"). -nh Locus names are supplied as first line of input file. -nf # Locus names are supplied in file #. This file should have one line per locus, with the locus name as the first field. -pr # The threshold for prior trimming is # (default is zero). -po # The threshold for posterior trimming is # (default is 0.01). -q "Quiet" operation. This suppresses the constantly-updated progress report that is written to the screen. This is needed when the program is to be run in "batch" mode. -ro Add new loci in random order (off by default). -rv Add new loci in reverse order (off by default). -rp When -mm option is set, each repeated EM algoritm is restarted with random values for the prior haplotype probbailities. If not set, each EM algorithm is restarted with random posterior assignment probabilities for each subject (the default behaviour). -rs Select random starting point for each EM iteration. Otherwise we start by assuming linkage equilibrium between the new marker and the previous ones (default is "off"). -sd # Set the pseudo-random number generator seed to a large integer, #. The default is to generate a seed from the date and time. -ss Specifies that the output file should be written as a tab-delimited file with variable names on the first line (suitable for reading into spreadsheet or statistical programs; default is "off"). -to # The convergence criterion for the EM iterations is # (the tolerated change in log-likelihood between two iterations; default 0.0001). -th # A number between 0 and 1, controlling the posterior threshold for writing most likely haplotype assignments to subjects to output-file-2. Only assignments whose posterior probability exceeds this multiple of the most likely a posterior assignment will be written to the output file (not relevant when in multiple imputation mode). Multiple imputation options: -mc # The number of MCMC steps between imputations -ds # The starting value of the df parameters for the Dirichlet prior. This is specified as a multiple of the number of chromosomes observed (i.e. twice the number of subjects). The default value is 0.1. -de # The final values of the df parameters for the Dirichlet prior (specified in the same way as above). The default value is zero, corresponding to complete ignorance. If the command is issued without options or arguments, a brief description of available options is written to the screen. Output -----1. Iteration progress reports (written to the screen). Note that some terminal emulators which provide "scrolling" may seriously slow down operation of the program. In this case you should either use a standard nonscrolling xterm, or invoke the -q option which suppresses this output. 2. A file listing the haplotypes found, and their probabilities (output-file-1). The list is in descending order of probability and a cumulative probability is also listed. The cumulative probability is suppressed if the -ss option is in force. 3. A file listing assignments of haplotypes to subjects (output-file-2). This file contains all assignments whose posterior probability exceeds a multiple of that of the most probable assignment (see -th option). The file is in "long format" -- that is, the pair of haplotypes for each assignment appear as successive lines. 4. A file (named "snphap-warnings") which contains any warning messages. Output files output-file-1 and output-file-2 are in a compressed and easily readable format. Alternativelly they can be saved as tab-delimited text files suitable for reading into a spreadsheet program, or a statistical program such as "Stata". Both file names are optional and a missing argument can be indicated with a single "." (period or full-stop) character. But since it must be assumed that you want SOME output, omission of both file names causes the program to default to "snphap.out" for output-file-1. In multiple imputation mode, an additional series of files is created. Each imputation causes a fresh file (or pair of files) to be written. The file names are as specified on the command line, but the strings .001, .002, .003 ... etc. are appended. Building -------- A primitive Makefile is supplied. This uses the gcc compiler and will need to be edited if a different C compiler is to be used. You may also need to edit the CMP_FLAGS and LD_FLAGS options (which provide flags used by the compiler at compile and load stages respectively) For Microsoft Windows users, I suggest use of emulation package. See the "Cygwin" Unix http://www.redhat.com/software/tools/cygwin I found that setting LD_FLAGS to -lm worked for me on both Linux and Solaris (this is the default setting), but on Cygwin I had to omit this flag. The default uniform random number generator (UNIFORM_RANDOM) is set to be the standard 48-bit function `drand48', and the corresponding seeding function (RANDOM_SEED) is `srand48'. However, for systems which do no support the 48-bit functions (this includes Cygwin), the 32-bit versions can be chosen: UNIFORM_RANDOM = drand RANDOM_SEED = srand `drand()' is defined as a macro evaluating to (0.5+rand())/(1+RAND_MAX). A short test data file is also included. This contains typings of 100 subjects for 51 SNPs in a small region. To test the program: ./snphap test.dat Altrenatively, if you wish to incorporate locus names in the output, ./snphap -nf test.nam test.dat Acknowledgements ---------------Thanks to Newton Morton and Nikolas Maniatis for their helpful comments and suggestions on an early previous version. Thanks also to anyone who has pointed out bugs in earlier versions. David Clayton Diabetes and Inflammation Laboratory Tel: 44 (0)1223 762669 Cambridge Institute for Medical Research Fax: 44 (0)1223 762102 Wellcome Trust/MRC Building david.clayton@cimr.cam.ac.uk Addenbrooke's Hospital, Cambridge, CB2 2XY wwwgene.cimr.cam.ac.uk/clayton