app39904-sup-0007-suppinfo2
advertisement

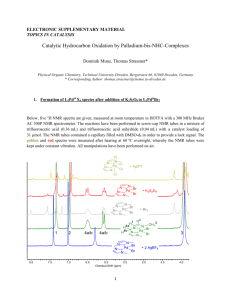
Electronic supplementary information (ESI) RESULTS AND DISCUSSION Synthesis and characterization of dihydroxy compounds containing ester groups In this study, three types of dihydroxy compounds each containing two ester groups were utilized, a dihydroxy compound containing aliphatic–aliphatic ester group (CH– Bu), a dihydroxy compound containing aromatic-aliphatic ester group (t-PHTH–Bu) and a dihydroxy compound containing aromatic-aromatic ester group (t-PHTH–BPA). The aliphatic-aliphatic and aromatic-aliphatic dihydroxy compounds were synthesized from the reaction of cis/trans-1,4-cyclohexanedicarboxylic acid dichloride (CHAC) or terephthaloyl chloride (t-PHTHC) with a 10 fold excess of 1,4-butanediol, respectively. The aromatic-aromatic dihydroxy compound (t-PHTH–BPA) was prepared by the reaction of terephthaloyl chloride with a 25% excess amount of the stoichiometric quantity of BPA according to a previously published procedure.1,2 Bis(4-hydroxybutyl) cis/trans-1,4-cyclohexanedicarboxylate (CH–BuD) is a new compound and was obtained in high yield as a light yellow viscous liquid. The other two dihydroxy compounds, t-PHTH–BuD4,5,6-11 and t-PHTH–BPA,1-3 were already known in the literature and were obtained as white solids in high yields. These dihydroxy compounds were characterized by melting point, FT–IR, 1H–NMR and 13 C–NMR spectroscopy. The yield, the physical properties and the labeling of the important IR bands of these dihydroxy compounds are summarized in Table S I. The melting point (Tm) of t-PHTH–BPA diol is 197–199°C (lit value is 199–201°C2) and the melting point of t-PHTH–BuD is 80°C (lit. value is 79–80ºC4,5). The melting point of t-PHTH–BPA diol was much higher than that of t-PHTH–BuD, this may be due to the extensive aromatic character of t-PHTH–BPA diol. TABLE S I FT–IR spectroscopy The FT-IR spectra of dihydroxy compounds showed strong and broad absorption bands from 3409 to 3426 cm-1 for the stretching vibrations of the alcoholic groups, strong absorption bands from 1710 to 1736 cm-1 for the C=O stretching frequency of the ester group, and from 1142 to 1282 cm-1 for the C–O–C stretching vibrations. These IR data are conformed to the reported literature.1,2,10,12 and thus suggest the formation of the postulated dihydroxy compounds containing ester groups. NMR spectroscopy 1 H-NMR spectra: The 1H−NMR spectra of the dihydroxy compounds showed peaks for all proton types present. The signal for the proton of the butyl hydroxyl group in CH–BuD was observed at δ = 3.06 and that in t-PHTH–BuD at δ = 2.71 ppm, respectively, while that of BPA in t-PHTH–BPA was observed at δ = 9.16 ppm. The signal of the terminal aliphatic methylene protons of Bu moiety attached to the hydroxyl group (–CH2OH) in both CH–BuD and t-PHTH–BuD were observed as triplet at δ = 3.50 and 3.63 ppm, respectively. The signals for the terminal aliphatic methylene protons of Bu moiety attached to the oxygen of the ester group (– COOCH2–) in CH–BuD and in t-PHTH–BuD were observed at δ = 3.96 and 4.29 ppm, respectively. The signal of the tertiary aliphatic CH proton in CH–BuD attached to the carbon of the C=O of the ester group (–CH–COO) was observed at δ = 1.91 ppm. The signals of the methylene protons of cis/trans-1,4-cyclohexylene ring and those of the central protons of Bu unit were observed as masked unresolved multiplets in the range δ =1.2–1.8 ppm. For the t-PHTH unit in t-PHTH–BuD and t-PHTH– BPA, the signal of the terephthalate ring protons appeared as singlet at δ = 7.99 ppm and at δ = 8.25 ppm, respectively. The signals of the aromatic protons of BPA unit near the ester group were observed in the range δ = 7.1–7.3 ppm, while those next to the alcoholic group were observed at δ = 6.65 ppm. The peak of the methyl protons of BPA units was observed at δ = 1.57 ppm. These 1H–NMR data are conformed to the reported literature2,5,8,10,11 and thus suggest the formation of the postulated dihydroxy compounds containing ester groups. 13 C-NMR spectra: In the 13C–NMR spectra of the dihydroxy compounds, the signals of the terminal methylene carbon atoms of Bu moiety attached to the hydroxyl group (–CH2OH) of both CH–BuD and t-PHTH–BuD were observed at about δ = 62 ppm. The signals of the terminal aliphatic methylene carbon atoms of Bu moiety attached to the oxygen of the ester group (–COOCH2–) in CH–BuD and in t-PHTH–BuD were observed at δ = 64.2 and 65.3 ppm, respectively. For cyclohexylene ring carbons of CH–BuD, the signal of the tertiary aliphatic carbon atom (CH) attached to the carbon of the C=O of the ester group (–CHCOO–) was observed at δ = 42.4 ppm. The signals for cyclohexylene ring methylene carbon atoms appeared at δ = 25–28 ppm. For the tPHTH unit of t-PHTH–BuD and t-PHTH–BPA, the signals of the quaternary aromatic ring carbon atoms next to the C=O of the ester groups appeared at δ = 134 ppm. The other aromatic ring carbon atoms of both t-PHTH–BuD and t-PHTH–BPA appeared at δ = 130 ppm. Interestingly, the signal of the quaternary carbon atoms of the aromatic rings of the BPA in t-PHTH–BPA attached to the hydroxyl groups appeared at a high δ value (δ = 156 ppm). Apparently, this peak (Table S II, entry 3, carbon 13) lies a way enough from in the carbonyl carbon signal region of the aromatic or aliphatic ester group and, therefore, does not mask with it. The signals of the carbon atoms of the ring in ortho– and meta– positions to the alcoholic group appeared in the range δ = 115 and 128 ppm, correspondingly. The signal of the quaternary aromatic carbon atoms in para position to the hydroxyl group which is attached to the aliphatic quaternary carbon atom appeared at δ = 148 ppm. The signal of the aliphatic quaternary carbon atom (bearing the methyl groups) appeared at δ = 42.0 ppm, while that of the methyl carbon atom appeared at δ = 31.2 ppm. The signal of the quaternary carbon atom of the aromatic ring attached to the oxygen of the ester group appeared at δ = 141 ppm. The signals of the carbon atoms of the ring in ortho– and meta– positions to the ester group appeared at δ = 122 and 128 ppm, respectively. Finally, the signal of the carbonyl carbon of the aromatic-aromatic ester of t-PHTH–BPA appeared at δ = 164 ppm, aromatic-aliphatic ester of t-PHTH–BuD at about 166 ppm and aliphatic-aliphatic ester (i.e., CH–BuD) at 175 ppm, which are the correct positions for the carbonyl carbon atoms of these ester. These 13 C–NMR data are conformed to the reported literature.1,2,5,8 and thus suggest the formation of the postulated dihydroxy compounds containing ester groups. The 1H–NMR and 13 C– NMR data for the various dihydroxy compounds prepared are reported in Table S II. TABLE S II Synthesis and characterization of monomers The prepared dihydroxy compounds containing ester groups were reacted with PCF1315 to obtain the following diphenyl dicarbonate monomers: bis(4-hydroxybutyl) cis/trans-1,4-cyclohexanedicarboxylate diphenyl dicarbonate (CH−Bu DPDC), bis(4hydroxybutyl) terephthalate diphenyl dicarbonate (t-PHTH−Bu DPDC) and 1,4-[di(4-hydroxy-diphenyl-2,2'-propane)] terephthalate diphenyl dicarbonate (t- PHTH−BPA DPDC). To our knowledge, all these monomers are novel compounds; therefore, they were characterized by melting point, FT–IR, 1H–NMR and 13 C–NMR spectroscopy to confirm their structures. Their structures, yield, physical properties and the labeling of the important IR bands are summarized in Table S II. FTIR spectroscopy The FT–IR spectra of the diphenyl dicarbonate monomers showed strong absorption bands due to the C=O stretching vibrations of the carbonate group from 1759 to 1778 cm-1 and strong absorption bands due to the C=O stretching vibration of the ester group from 1720 to 1739 cm-1. The FT–IR spectra also showed that the C=O stretching frequency of aromatic-aromatic carbonate group (e.g., t-PHTH–BPA DPDC) was the highest among the monomers. The absorption bands of the C–O–C stretching vibrations were observed in the range from 1187 to 1253 cm-1. The yield, melting points and the labeling of the important IR bands of monomers are summarized in Table S I. NMR spectroscopy 1 H–NMR spectra: In the 1H–NMR spectra of CH–Bu DPDC and t-PHTH–Bu DPDC monomers, the signals of the terminal methylene protons of Bu moiety attached to the ester group (–COOCH2–) were observed as triplets at δ = 4.11 and 4.32 ppm, respectively, while those next to them (–COOCH2CH2–) in CH–Bu DPDC were masked with CH ring protons, and in t-PHTH–Bu DPDC were observed at δ = 1.92 ppm. In the spectra of CH-Bu DPDC and t-PHTH–Bu DPDC monomers, the signals of the terminal aliphatic methylene protons of Bu moiety attached to the carbonate group (–OCOOCH2–) were observed as triplets at δ = 4.25 and 4.40 ppm, respectively, while those next to them (–COOCH2CH2–) in CH–Bu DPDC were masked with CH ring protons, and in t-PHTH–Bu DPDC were also observed at δ = 1.92 ppm. In the spectrum of CH–Bu DPDC, the signal of the tertiary aliphatic (CH) protons attached to the carbon of the C=O of the ester group (–CH–COO–) was observed at δ = 2.03 ppm, the signal of cyclohexylene ring methylene protons were observed in the range δ = 1.3–1.9 ppm. In the spectrum of t-PHTH–Bu DPDC, the signal of the aromatic terphthalate ring protons was observed at δ = 8.09 ppm. Regarding the spectrum of t-PHTH–BPA DPDC the signal of the terephthalate aromatic ring protons was observed at δ = 8.30 ppm, while the signals of BPA aromatic ring protons were observed in the range δ = 7.1–7.5 ppm. The signal for the methyl protons of BPA unit were observed at δ = 1.70 ppm. On the other hand, the signals of the phenyl ring of phenyl carbonate group ortho, meta and para to the carbonate group were observed in all monomers around δ = 7.15, 7.38 and 7.25 ppm, correspondingly. 13 C–NMR spectra: In the 13 C–NMR spectra of CH–Bu DPDC and t-PHTH–Bu DPDC, the signal for the terminal methylene carbon atoms of Bu attached to the ester group (–COOCH2–) appeared at δ = 63.7 and 64.8 ppm, respectively, while that next to them of CH–Bu DPDC (–COOCH2CH2–) appeared in the range δ = 25–29 ppm and that of t-PHTH–Bu DPDC at δ = 25.2 ppm. The signal for the methylene carbon atoms of Bu attached to the carbonate group (–OOCOCH2–) were observed at δ = 68.2 ppm for both monomers, while that next to them (–OOCOCH2CH2–) in CH–Bu DPDC was observed in the range δ = 25–29 ppm and that in t-PHTH–Bu DPDC at δ = 25.5 ppm. In the spectrum of CH–Bu DPDC, the signal of the tertiary aliphatic (CH) carbon atom attached to the C=O of the ester group (-CH–COO) was observed at δ = 42.5 ppm and the signals of cyclohexylene ring methylene carbons at δ = 25–29 ppm. In the spectrum of t-PHTH–Bu DPDC, the signal of the aromatic t-PHTH ring carbons appeared at δ = 130 ppm and the signal of the quaternary aromatic ring carbon attached to the C=O of the ester appeared at δ = 134 ppm. In the 13 C–NMR spectra of CH-Bu DPDC, t-PHTH–Bu DPDC and t-PHTH–BPA DPDC, the signal for the aliphatic-aliphatic, aromatic-aliphatic and aromatic-aromatic C=O of the ester group appeared at δ = 175.0, 165.8 and 164.4 ppm, correspondingly. The corresponding signals of the carbonate groups of these monomers appeared at δ = 153.7, 153.8 and 152.2 ppm, respectively. Regarding the spectrum of t-PHTH–BPA DPDC the signal of the terphthalate aromatic ring carbon atoms appeared at δ = 130 ppm. The quaternary aromatic carbon of the terephthalate unit attached to C=O of ester appeared at δ = 134 ppm, while that of BPA unit attached to the oxygen of the ester appeared at 149 ppm. The peaks of the aromatic carbon atoms of BPA ortho and meta to the ester group appeared at δ = 121 and 128 ppm, respectively. The signals of the quaternary carbon atoms of the aromatic rings next to the aliphatic quaternary carbon appeared at about 148 ppm. The signal of the aliphatic quaternary carbon atom bearing the methyl groups appeared at δ = 42.7 ppm, while the signal of the methyl carbon atoms appeared as at 31.0 ppm. The signals of the quaternary carbon atoms of the aromatic rings of BPA next to the carbonate groups appeared at δ = 151 ppm. The signals of the carbon atoms of the ring in ortho and meta positions to the carbonate group appeared at δ = 121 to 128 ppm. On the other hand, the quaternary carbon atom of the phenyl ring of the phenyl carbonate group of the three monomers attached to carbonate group appeared at δ = 151 ppm. The carbon peaks of the phenyl ring ortho, meta and para to the carbonate group in all monomers appeared at δ = 121, 129 and 126 ppm, correspondingly. The 1H–NMR and 13 C–NMR spectral data assigned to protons and carbons of the various monomers are presented in Table S II. Synthesis and characterization of poly(ester carbonate)s FT–IR spectroscopy The poly(ester carbonate)s prepared were analyzed by FT–IR. The IR spectra showed two strong absorption bands due to the C=O stretching vibration; one for the carbonate group and the other for the ester group. The spectra showed that the C=O stretching frequencies of aromatic-aromatic polycarbonates were higher than those of aromatic-aliphatic polymers which is in turn higher than those of aliphatic-aliphatic polymers. The spectra showed strong absorption band due to the C=O stretching vibration of the ester group around 1740 cm-1 for aromatic-aromatic ester, around 1720 cm-1 for aromatic-aliphatic ester and around 1730 cm-1 for aliphatic-aliphatic ester. The absorption bands for aromatic-aromatic carbonate were above 1770 cm-1, for aromatic-aliphatic carbonate around 1760 cm-1 and for aliphatic-aliphatic carbonate below 1750 cm-1. Strong absorption bands for the C–O–C stretching vibration for all polymers were observed in the range 1164 to 1270 cm-1. These IR data, which are typical for the carbonate and ester groups, are in accordance with the data reported in literature16,17 and, therefore, confirm the formation of the various poly(ester carbonate)s in this work. NMR spectroscopy 1 H–NMR spectra: The proton NMR spectra of aromatic-aliphatic and aliphatic- aliphatic poly(ester carbonate)s confirm the formation of the polymers. The chemical structures of the various polymers in all series essentially consisted of CH–Bu, tPHTH–Bu and t-PHTH–BPA units bonded to different alkylene or arylene units of the dihydroxy compounds by a carbonate group. In their 1H–NMR spectra, the polycarbonates of each series showed similar pattern of peaks for each of these units, except for the aliphatic or the aromatic dihydroxy compound moieties attached to them. The terminal methylene protons of Bu unit attached to the oxygen of the carbonate group (–OOCOCH2–) in CH–Bu, t-PHTH–Bu units showed down field effects, and their signals in polycarbonates was observed as multiplet in the range from δ = 4.10 to 4.40 ppm, the signals of the methylene protons next to them appeared as multiplet in the range from δ = 1.78 to 1.94 ppm. The signal of the terminal methylene protons of Bu unit attached to the oxygen of the ester group (– COOCH2–) was observed as multiplet in the range from δ = 4.03 to 4.33 ppm, the signals of the methylene protons next to them appeared as multiplet in the range from δ = 1.63 to 1.86 ppm. For poly(ester carbonate)s containing cyclohexylene units, the signals due to cyclohexylene ring protons appeared as multiplets in the range δ = 1.2– 1.9 ppm. For poly(ester carbonate)s containing terephthalate units, the signal of the aromatic protons appeared in t-PHTH–Bu and t-PHTH–BPA units as singlet at about δ = 8.07 and 8.31 ppm, respectively. The signals of the other aromatic protons (BPA, BIPH, HQ) of the diols employed appeared in the range δ = 7.0–7.7 ppm. The signals of the aliphatic protons of Bu unit of BuD next to the carbonate group and that of those next to them were observed at about δ = 4.40 and 1.86 ppm. The signals of the aliphatic protons of DIGOL unit of the dihydroxy compound next to the carbonate group were observed in the range δ = 4.03–4.40 ppm and those next to the ethereal oxygen were observed in the range δ = 3.62–3.82 ppm, respectively. On the other hand, the 1 H–NMR spectra of aromatic-aromatic poly(ester carbonate)s were generally not indicative of formation of these polycarbonates. The signals of all protons were collectively observed as asymmetric multiplets in the aromatic region of the spectra. 13 C–NMR spectra: The 13 C–NMR spectra of poly(ester carbonate)s were quite informative and showed signals due to all aliphatic carbon types. The polymers synthesized exhibit a close similarity in the core structure, derived from the diphenyl dicarbonate monomers, but differs in the alkylene or the arylene parts of the various dihydroxy compounds. Therefore, the change in the pattern of 13 C–NMR spectra of polymers could be correlated to the structure of the dihydroxy compounds. With the exception of the alkylene and arylene moieties of the dihydroxy compounds, the patterns of the 13C–NMR spectra for the CH–Bu unit, t-PHTH–Bu unit, and t-PHTH– BPA unit, in all polycarbonates series were similar to those of their monomers. The positions of the aliphatic and aromatic carbon signals of these units in the spectra of all polymers containing these units were observed at chemical shifts close to those in the monomers. All 1H–NMR and 13 C–NMR peaks were assigned to protons and carbons of the various polymers synthesized in Series A, B, and C and their assignments are presented in Tables S III, S IV and S V, respectively. Table S III Table S IV Table S V The 1H–NMR and 13 C–NMR signals of the terminal methylene groups of all polycarbonates attached to the carbonate group were highly indicative for the formation of the postulated polycarbonates. The attachment of the terminal methylene group to the oxygen of the carbonate led to a downfield shift, which was reflected in the chemical shifts in the proton spectra of the polycarbonates in the range δ = 4.10– 4.40 ppm and carbon spectra in the range, and δ = 67.4–69.1 ppm. This effect was highly distinguished since the proton and carbon signals of the methylene carbon attached to the ester or to the ethereal oxygen (–CH2OCH2–) appeared at smaller δ values. This effect represents a strong spectral evidence of the formation of the various poly(ester carbonate)s in this work. For example, this result was typically demonstrated by the 1H–NMR spectra of Series B polymers, which showed peaks for terminal proton signals at δ = 4.35–4.40 ppm attached to the carbonate group, at δ = 4.15–4.33 ppm attached to the ester group and at δ = 3.81 ppm attached to the ether group. The corresponding peaks of the carbon atoms attached to the carbonate group, ester group and ether group are less conformed to this trend and appeared at about δ = 68 ppm, 65 ppm and 67 ppm, correspondingly. REFERENCES 1. Lakshmi M. S.; Reddy, B.S.R. Eur. Polym. J. 2002, 38, 795. 2. Abd-El-Aziz, A. S.; Todd, E. K.; Ma, G. Z. J. Polym. Sci. Part A: Polym. Chem. 2001, 39, 1216. 3. Lakshmi, M. S.; Srividhya, M.; Reddy, B. S. R. J. Appl. Polym. Sci. 2003, 88, 2963. 4. Lyoo, W. S.; Kim J. H.; Ha, W. S. J. Appl. Polym. Sci. 1996, 62, 473. 5. Biemond, G. J. E.; Brasspenning, K.; Gaymans R. J. J. Appl. Polym. Sci. 2012, 124, 1302. 6. Kim, J. H.; Park, J. H.; Jang, H. K.; Yoon, J. Y.; Lyoo, W. S. J. Appl. Polym. Sci. 2003, 90, 2200. 7. Kim, J. H; Park, J. H.; Kwon, C. H.; Lyoo, W. S. J. Polym. Sci.: Part A: Polym. Chem. 2002, 40, 2435. 8. Mansour, S. H.; Ikladious, N. E. Polymer Testing 2002, 21, 497. 9. Sivaram, S.; Upadhyay, V. K.; Bhardwaj, I. S. Polymer Bulletin (Berlin, Germany) 1981, 5, 159. 10. Padias, A. B.; Hall, H. K. J. Polym. Sci., Polym. Chem. Edition 1981 19, 1021. 11. Shaik, A. A.; Richter, M.; Kricheldorf, H. R.; Krüger, R-P. J. Polym. Sci.: Part A: Polym. Chem. 2001, 39, 3371. 12. Gillette, P. C.; Dirlikov, S. D.; Koenig, J. L.; Lando, J. Polymer 1982, 23, 1759. 13. Liaw, D–J.; Chang, P. Polymer 1996, 37, 2857. 14. Sun, S. –J.; Chang, T.-C. J. Polym. Sci. Part A: Polym. Chem. 1993, 31, 2237. 15. Sun, S. –J.; Chang, T. C.; Li, C. H. Eur. Polym. J. 1993, 29, 951. 16. Hait, S. B.; Sivaram, S. Macromol. Chem. Phys. 1998, 199, 2689. 17. Shaikh, A. G.; Sivaram, S. Polymer 1995, 36, 3223. B.,