Principle of purification strategies
advertisement

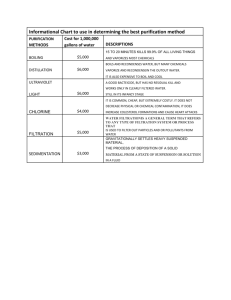
Thida Chanyachukul Introduction The development of techniques and methods for the separation and purification of product molecules have been an important prerequisite for many of the advancements made in pharmaceutical manufacture. Improvements in materials and utilization of modern instruments have made separation more predictable and controllable. However, not all problems in purification system are solved by the acquisition of sophisticated laboratory equipment and column packing, which give high selectivity and efficiency. Difficulties still remain in finding optimum conditions and choosing suitable methods. Our interest is focused on improvement of AIDS drugs (such as protease inhibitors, HIV vaccine and IL-2), and then great care should be taken in the selection of suitable techniques to obtain each efficient product. I will overview first the different purification techniques and their principles of operation. In the subsequent part some more detail in each purification work on all three types of the drugs will be discussed. After that I will conclude with ideal and suitable limits in purity degree. Purification strategies Today it is possible to base the purification of a pharmaceutical product on the knowledge of its molecular properties, structural as well as function. Suggestions on how to solve a purification problem can best be made if data on its structure and function, including particular structural details, is available. Conversely, results from the application of a particular purification method can often be interpreted in terms of molecular properties, often in a detailed manner. Production can be divided into “upstream” and “downstream” processing. As defined here, upstream processing refers to the initial fermentation process, which results in the initial generation of product. Downstream processing refers to the actual purification of the product and generation of finished product format (i.e. filling into its final product containers, freeze-drying if a dried product format is required) followed by sealing of the final product containers. Subsequent labeling and packaging steps represents the very final steps of finished product manufacture.1The quality and biosafety of a pharmaceutical is to a great extent, dependent on the extraction procedures used to manufacture the purified product. On the one hand, down stream processing has to ensure an effective and economic isolation of the desired product from the culture broth or cellular material obtained during the cell culture process. However, components that would contaminate the final product must be reliably separated. Different types of components that should not be present in the final product formulation have to be removed during these steps. These can be classified into two groups as in table 1. The removal of medium components and proteinaceous impurities is an integral part of product isolation. Nevertheless, the removal of medium supplements, such as antibiotics or cytotoxic substances must be guaranteed by the purification strategy and appropriate tests have to be established to validate their efficiency.5 1 Thida Chanyachukul Table1: Categories of components that should not be present in the final Pharmaceutical product Components present due to process conditions : - Host-cell-derived components : DNA and proteins - Process-derived components : lipid, proteins, antifoam agents, antibiotics, substances used for product isolation, cleansing agents Components present due to contamination : - Adventitious agent : viruses, virus-like particles, bacteria, fungi, mycoplasmas, transmissible spongiform encephalopathy agents Because, for practical reasons, it is not possible to manufacture a 100% pure product, acceptable concentration levels for the presence of impurities in the final product formulation have been defined. For example, WHO defined the maximal acceptable amount of DNA to be 100 pg per single dose of a biotechnological derived protein drug.4 Working on reaction volumes of between 2-5 liters usually requires the use of pilot-plant equipment. Many purification techniques are not practical when dealing with large quantities of material. In general the most useful methods of purification that can be applied to large quantities of material are recrystallization for solids, and distillation or steam distillation for liquids. Chromatography should be avoided if possible since it becomes a very expensive operation on large scale, but if it is necessary, then medium pressure liquid chromatography (mplc) is the method of choice. Principle of purification strategies Basic concept of Filtration6 The usual technique is to pass the solution, cold or hot, through a fluted filter paper in a conical glass funnel. It is used to remove particulate impurities from liquids or to collect insoluble or crystalline solids, which crystallize, from solution. When the solid particles are too fine to be collected on a filter funnel because filtration is extremely slow, separation by centrifugation should be used. Basic concept of Gel filtration2 The separation may simply be regarded as due to the different amount of time different solute stay within the liquid phase that is entrapped by the matrix. This time is of course related to the fraction of the pores that is accessible to the solute. The interpretation of this fraction in terms of pore dimension and gel structure, together 2 Thida Chanyachukul with various expressions for solute size, results in slightly different equations for relating the distribution coefficient to the size of the solute. Ultrafiltration separates molecules on the basis of size and shape9 Filtration devices used in industry 7 Even though gel filtration is an uncomplicated and straightforward technique, there are some points worth consideration before starting the experimental work. The actual sample may require a special pH, solvent, additives or pretreatment to yield a true solution. The next step is to select the gel that will cope with the chosen solvent and pH and that has a suitable separation range. Possible adsorption properties of the gel must also be considered. The nature of the separation and the sample may put demands on such parameters as resolution, separation time and sample load which in turn are partly dependent on selected gel. The choice of column dimensions and the packing efficiency of the column however, also affect these parameters. Obviously, for different separation problems, such as desalting, preparative purifications or analytical separations different requirements should be stressed. Economic factors and the possibilities of scaling up may also are important. Basic concept of chromatography2 The term chromatography refers to a group of separation techniques, which are characterized by a distribution of the molecules to be separated between two phases, one stationary and the other mobile. Molecules with a high tendency to stay in the stationary phase will move through the system at a lower velocity than will those which favor the mobile phase. The shape, rigidity and particle size distribution profile of the gel matrix are important parameters, which govern the performance of the stationary phase. Chromatographic techniques for analysis and purification of reaction products are probably the most universally important of all the skills in which an organic chemist requires expertise. Various chromatographic techniques which may be used to separate proteins from each other. The basis upon which separation is achieved is also listed in table 22 3 Thida Chanyachukul Table 2 : Chromatographic technique Ion exchange Basis of separation Differences in protein surface charge at a given pH Differences in size/shape of different proteins Differences in the size and extent of hydrophobic patches on the surface of proteins Ability of a protein to bind in a biospecific manner to a Chosen immobilized ligand Immobilized antibodies may be used as affinity absorbents for the antigen that stimulated their production Purification of proteins which displays ability to interact tightly with selected dyes Ability of certain proteins to complex with zinc and copper Mechanism not fully understood. Involve ability of some proteins to Bind to calcium and phosphorus on the surface of hydroxyappetite crystals Gel filtration Hydrophobic interaction chromatography Affinity chromatography - Immunoaffinity chromatography - Dye affinity chromatography Metal chelate chromatography Hydroxyappetite chromatography A variety of different chromatographic techniques are available which separate molecules from each other on the basis of differences in various physiochemical characteristics. In general, a combination of two to four different chromatographic techniques is employed in a typical downstream processing procedure. Gel filtration and Ion exchange chromatography are the among the most common. Affinity chromatography is employed wherever possible as its high biospecificity facilitates the achievement of a very high degree of purification. There are often times when quite difficult separations need to be performed on a fairly large scale. Even if the mixture will separate by flash chromatography, it may be prohibitively expensive, especially if it is a step, which needs to be carried out routinely. This is one occasion when mplc is very useful. The resolution of mplc is somewhat better than flash chromatography, but another important feature is that the columns are reused many times, thus avoiding the expense of throwing away large quantities of silica.8 The mplc system is essentially a simplified and much cheaper version of an hplc set-up. At the heart of the system is any type of pump, which will operate at 100 psi, with a controllable flow rate of up to 100 ml/min. The sample introduced into the system via an injection valve, but this is a much simpler and less expensive valve than that found in an hplc system. There is a choice of silica available for mplc, but for 4 Thida Chanyachukul most purposes ordinary “flash” (40-60 um) silica is used. A column packed with 1520 um silica is more effective for difficult separations but there is a lot to be said for sticking for only one, or perhaps two types of silica and becoming familiar with their characteristics. In most cases getting a good separation is simply a matter of choosing the appropriate size of column and the correct solvent. Other solid phases can also be used on a mplc system alumina, ion exchange resin, Sephadex.8 mplc (medium chromatography) pressure liquid 8 It is very useful to have the mplc system linked to a fraction collector. An UV or refractive index detector can also be incorporated into the system, or TLC can simply analyze the fractions, as for flash chromatography. If the conditions are kept constant the results from mplc are very consistent, so if you wish to repeat separation of the same mixture, it is very easy to predict which fractions will contain each component. And to define which fraction contains Ritonavir, you should have to analyze them again by affinity chromatography or mass spectroscopy. Basic concept of Ion exchange chromatography (IEC)2 Protein charge properties are used for fractionation purposes in several techniques. Thus, electrophoresis depends on electrophoretic mobility, which in turn is a function of charge density, while isoelectric focusing separates proteins according to their isoelectric points, i.e. the pH of zero net charge. IEC not only depends on charge density, but also on the distribution of charges on the protein surface, i.e. charge anisotropy. Similarly separation in chromatofocusing reflects not only the pI of a protein, but also on the shape of its titration curve in the vicinity of the pI. The reason for the popularity of IEC is its versatility, its high resolving power, its high capacity and its straightforward basic principle. Principle of chromatography2 5 ion exchange Thida Chanyachukul The basis for the IEC process is the competitive binding of ions of one kind, in our case proteins, for ions of another kind, for example other proteins or salt ions of the same charge, to an oppositely charged chromatographic medium, the ion exchanger. The interaction between the proteins and the ion exchanger depends on several factors: net charge and surface charge distribution of the protein; the ionic strength and the nature of the particular ions in the solvent; pH, or strictly speaking in the proton activity; and other additives to the solvent, such as organic solvents etc. It is clear that the more highly charged a protein is, the more strongly it will bind to a given, oppositely charged ion exchanger. Similarly, more highly charged ion exchangers, i.e. those with a higher degree of substitution with charged groups, bind proteins more effectively than weakly charged ones. Conditions, for example pH, which alter the effective charge on either the protein or the ion exchanger will affect their interaction and may be used to influence the ion exchange process. The term strong and weak ion exchanger derive from the pKa’s of their charged groups and do not say anything about the strength with which they bind proteins. At pH’s far from the pKa, binding will be equally strong to either a weak or a strong ion exchanger. IEC is eminently well suited to purification of either large quantity of proteins, because of its high loading capacity or purification from large volumes of sample, because of its ability to concentrate proteins from dilute solution. Although process scale IEC is routinely carried out with column as large as 170 liters, even a standard laboratory column with a volume of 500 ml is usually sufficient to concentrate and purify 5 grams of protein from as much as 10 liters of a dilute sample. In practical terms, large scale IEC differs mainly in the increased use of step, as opposed to continuous gradient, elution procedures. Otherwise exactly the same general procedures and principles are used as in a smaller scale. It is advisable to obtain this increase in volume by choosing a column with a larger diameter to avoid excessively long separation times. The multiple inlet design is suitable. The choice of buffer systems for large scale work will naturally be influenced by cost considerations, but the needs for adequate buffering capacity remain and should not be sacrificed for small cost savings. Basic concept of affinity chromatography2 Affinity chromatography owes its name to the exploitation of these various biological affinities for adsorption to a solid phase. One of the members of the pair in the interaction, the ligand, is immobilized on the solid phase, while the other, the counterligand (most often a protein), is adsorbed from the extract that is passing the chromatographic column as you can see in table 3. A typical separation by affinity chromatography consists of four stages: adsorption, washing, elution and column regeneration. In many cases affinity chromatography is a very powerful method. This is particularly true when the product of interest is a minor component of a complex mixture. A property that needs special consideration is the association strength, between ligand and counterligand. If it is too weak there will be no adsorption, if it is too strong it will be difficult to elute the product adsorbed. It is always important to find conditions, such as pH, salt concentration or inclusion of, e.g., detergent or other 6 Thida Chanyachukul substances, that promote the dissociation of the complex without destroying the active compound at the same time. Table 3 : Ligand Antibody Enzyme Lectin Nucleic acid Hormone, vitamin Sugar Counterligand antigen, virus, cell substrate analogue, inhibitor, co-factor polysaccharide, glycoprotein, cell surface receptor, membrane protein, cell nucleic acid-binding protein (enzyme or histone) receptor, carrier protein lectin, enzyme or other sugar binding protein Basic concept of distillation 6, 7 The distillation process involves boiling a liquid, condensing the vapors, and directing the resulting liquid into another vessel. The liquid is heated to its boiling point, at which point its vapor pressure equals the atmospheric pressure. When the apparatus is at equilibrium and the distillation is occuring.7 Almost without exception, this method can be assumed to be suitable for all organic liquids and most of the low-melting organic solids. The efficiency of a distillation apparatus used for purification of liquids depends on the difference in boiling points of the pure material and its impurities.6 The conventional apparatus for simple distillation 8 Basic concept of Extraction7 Distillation is somewhat wasteful process. There is always material of intermediate boiling point that represents a process loss. Extraction tends to be more quantitative and requires a long time because of tendency for emulsification. Laboratory studies to improve the speed of separation should be carried out. Solvents used may have to be changed or temperature adjusted, or a preliminary purification may have to be carried out by another method. 7 Thida Chanyachukul Basic concept of crystallization or recrystallization The most commonly used procedure for the purification of a solid material by crystallization from a solution involves the following steps: 6 (a) The impure material is dissolved in a suitable solvent, by shaking or vigorous stirring, at or near the boiling point, to form a near-saturated solution. (b) The hot solution is filtered to remove any insoluble particles. (c) It is then allowed to cool so that the dissolved substance crystallizes out. The rate of cooling has to be controlled to obtain the crystal size desired. Fast cooling will cause the generation of many nuclei, producing a large number of small crystals rather than the growth in size of those already formed. Also, fast cooling will cause inclusion of by-products in the crystals.7 (d) The crystals are separated from the mother liquor, either by centrifuging or by filtering, under suction, etc. Usually, centrifuging is much to be preferred because of the much greater ease and efficiency of separating crystals and mother liquor, and also because of the saving of time and effort, particularly when very small crystals are formed or when there is entrainment of solvent.7 (e) They are washed free of mother liquor with a little fresh cold solvent, then dried. Medium to large scale crystallization (> 1 g) a set-up along the line 8 Crystallization for purification should be carried out only as a last step. The crystallization process gives a yield of about 90% at best. A 10% yield loss is quite high. Of course, the material in the mother liquor can be purified with additional processing which are much more quantitative such as extraction or chromatography. 7 Plant-scale purification of vaccine and IL-2 protein During the cultivation process, the cells either secrete the desired protein into the culture medium or the product accumulates in the cells. In either case, the first step of the purification procedure is the separation of cells and cell debris. This is achieved by centrifugation or microfiltration techniques. The efficacy of this step is influenced by the viability of the cells and by the medium composition. The productcontaining fraction (either by culture broth or a crude cell extract) is then concentrated by ultrafiltration or diafiltration, precipitation, high-affinity absorption or extraction steps, which reduce the volume and prepare the material for the chromatographic 8 Thida Chanyachukul steps used for the final product purification.3 This diagram overviews a generalized downstream processing procedure employed to produce a finished-protein product biopharmaceutical (vaccine or IL-2) 3 Fermentation If protein is expressed Intracellularly if protein is expressed extracellularly Recovery of producer cells (centrifugation or filtration) Removal of cells from media (centrifugation or filtration) cellular disruption (homogenization) removal of cellular debris (centrifugation or filtration) initial purification / concentration concentration of containing(ultrafiltration / ion exchange or extracellular (ultrafiltration or precipitation) precipitation) productmedia chromatographic purification; usually 2-4 chromatographic step Adjustment of potency and addition of excipients sterile filtration and aseptic filling freeze-drying powder preparation liquid preparation sealing of final product container, labeling and packing 9 Thida Chanyachukul Plant-scale purification of Ritonavir silica gel + chromatography 2-aminoaldehyde diols (white solid) bromoacetate (white solid) precipitation / filtration silica gel chromatography diamine (white solid) filtration compound X (white solid) epoxide (white solid) distillation silica gel chromatography resin compound compound XXXIIIa Hydrophobic Interaction chromatography Ritonavir distillation Affinity chromatography filtration RITONAVIR (final product) CRYSTALLIZATION LABELLING AND PACKAGING The purification steps presented in the above plant scale synthesis are designed in a modulate fashion, but the full processes are listed in the section of designing plant (Kanathip’s report). In the full processes, informations from the patent 10 number 5846987 and simulation of superpro program are documented. In general, however, 10 Thida Chanyachukul industry tends to avoid hydrophobic and affinity chromatography even they are conceptually the best method to specify and separate Ritonavir from other organic molecules which has the same molecular property. (Affinity chromatography will use protease enzyme to coat in the column, then pass the solution through, if it contains Ritonavir after eluting you will get it and use mass spectroscopy to define again, MW is approximately 720). Because both are very expensive and complicated, it is not practical in making protease enzyme in the plant scale level. To elute out the product from chromatography, we adjust pH, change ionic strength (isocratic or gradient), use another more potent hydrophobic interaction or other potent substrate analogue. Crystallization may be repeated until the substance has a constant melting point or absorption spectrum, and a substance may be redistillated in a fractionating column until it distils repeatedly within a narrow, specified, temperature range. Figure 1 shows result of the simulation on Superpro designer for base case of 308 kg/batch. Noted that the reaction 1, 2, 4, 5 and 7 contribute to less than 10% but the reaction 3, 6, 8 and 9 contribute to more than 90% of total equipment cost for purification in base case as seen in Figure 1. Base case : 308 kg/batch Total equipment cost : 16,562 M$ Purification equipment cost : 7,505 M$ Fig1 : Purification cost based on each reaction rxn8 rxn7 8% rxn9 rxn1 rxn2 7% 1% 1% 3% rxn3 42% rxn6 35% rxn5 rxn4 1% 2% 11 Thida Chanyachukul Sensitivity analysis on the plant throughput (kg/batch) (Figure 2) shows that reaction 3 and 6 dramatically increase in the equipment cost for purification when the throughput increases. Whereas the reaction 8 and 9 are not too much sensitive to the throughput. total purification equipment cost ($US) Fig. 2 : Sensitivity analysis 18000000 16000000 14000000 77 kg/batch 12000000 154 kg/batch 10000000 308 kg/batch 8000000 618 kg/batch 6000000 928 kg/batch 4000000 2000000 0 rxn3 rxn6 rxn8 rxn9 reaction When we compare the equipment cost for purification to the total capital cost you can obviously see from Figure 3 that when the throughput increases, the equipment cost for purification and total fixed cost also increase. But the rate of increase of both costs is not the same, as the total fixed cost is increasing sharper than purification cost suggesting that the higher throughput, the less equipment cost for purification you have to invest compared to the total equipment cost. Fig 3 : Cost analysis based on different yields 70000000 60000000 50000000 40000000 30000000 20000000 10000000 0 77 kg/batch 154 kg/batch 308 kg/batch purification 618kg/batch 928 kg/batch total fixed cost 12 Thida Chanyachukul Conclusion Purity is a matter of degree. All commercial available chemical substances are in some measure impure during manufacture. The important question, then, is not whether a substance is pure but whether a particular sample is sufficient pure for some intended purpose. By suitable manipulations it is often possible to reduce levels of impurities to acceptable limits, but absolute purity is an ideal which can never be shown to be attained. When setting out to purify a chemical, it is desirable that the starting material should be of the grade commercially available. In general, at least two different methods should be used in order to ensure maximum purification. Things that should be looked into more detail are the time necessary to carry out each purification step and the variation of product yield. For example, the cost of operating the plant will go up if each step takes a long time. Reaction 1, 2, 4, 5 and 7 contribute to less than 10% whereas reaction 3, 6, 8 and 9 contribute to more than 90% of total equipment cost for purification. Sensitivity analysis states that reaction 3 and 6 dramatically increase in total equipment cost for purification when throughput increases. But the reaction 8 and 9 are not too much sensitive when throughput increase. In case of different throughput , it can be suggested that the more you produce, the less you invest for purification operation. The decision to market the product in liquid or powder form must be determined experimentally, as there is no way to predict the outcome for any particular material. Some may remain stable for month- or even year- in solution, particularly if stabilizing excipients are added and the solution is refrigerated. Other, particularly when purified, may retain biochemical activity for only a matter of hours or days when in aqueous solution. References 1. Berthold, W. and Walter, J. Protein purification: aspects of processes for pharmaceutical products. Biologicals 1994;22:135-150. 2. Janson JC. And Ryden L. Protein purification; principles, high-resolution methods, and application: VCH publishers, Inc. 1989. 3. Hesse F. and Wagner R. Developments and improvements in the manufacturing of human therapeutics with mammalian cell cultures. Trends in Biotechnology. 2000 ;18(4):173-180 4. WHO study group. Acceptability of cell substrates for production of biologicals. WHO technical report series; 1987: 747, 1-29. 5. Walter, J and Allgaier, H. Validation of downstream processes. In: Mammalian Cell Biotechnology in Protein Product.1997; 453-48. 6. Perrin DD., Armarego WLF. and Perrin DR. Purification of laboratory Chemicals. 2nd ed. : Pergamon Press. 1980. 13 Thida Chanyachukul 7. White HL. Introduction to industrial chemistry : John Wiley & Sons, Inc. 1986. 8. Leonard J., Lygo B. and Procter G. Advanced practical organic chemistry. 2 nd ed. Blackie academic & Professional. 1996. 9. Walsh G. Biopharmaceuticals : Biochemistry and Biotechnology. John Wiley & Sons, Ltd. 1998. 10. Kampf DJ. Patent number 5846987. 8 Dec. 1998. 14