Membrane proteins and drug discovery: what is the link

Drug Discovery Today: Informatics (Computational Drug Discovery)
1
2
3
4
5
6
7
8
9
Computational Analysis of Membrane Proteins: the Largest Type of Drug Targets
Yalini Arinaminpathy*
,1
, Donald M. Engelman
1
, Mark B. Gerstein
1
Address:
1
Department of Molecular Biophysics and Biochemistry, Yale University, 266 Whitney
10 Avenue, New Haven, CT 06520-8114, USA
11
12 * Corre s ponding Author:
13 Dr Yalini Arinaminpathy; E-mail: yalini.pathy@yale.edu
14
15
16 25 – 30 Word Teaser : The sheer pharmacological importance of membrane proteins
17 contrasts starkly with their limited number of known structures. Computational tools are
18 valuable in bridging the gap between their structure, function and mechanism.
19 Keywords: Membrane Proteins, Molecular Dynamics, KcsA, GPCR, ion channels.
1
Drug Discovery Today: Informatics (Computational Drug Discovery)
20 Abstract
21 The biological importance of integral membrane proteins, from the level of the
22 cell to entire organism is highlighted by the profound effects of various genetic
23 deficiencies in transporters and channels. Given their key roles and significance, it is
24 therefore necessary to understand their structures and thence mechanisms and regulation,
25 at the molecular level. Membrane proteins represent approximately 30% of currently
26 sequenced genomes. Paradoxically, however, at present, > 45,000 crystal structures are
27 deposited in the protein data bank (PDB), of which only 2% are of membrane proteins.
28 Interestingly, 971 protein folds are currently known, of which only 5% (49 folds) are of
29 membrane proteins. The great disparity between our understanding of soluble proteins
30 and membrane proteins has occurred largely due to the many practical problems of
31 working with membrane proteins, specifically difficulties in expression, purification and
32 crystallization. Thus, technological developments have been increasingly utilized in order
33 to make crucial advances in understanding membrane protein structure and function.
34
2
Drug Discovery Today: Informatics (Computational Drug Discovery)
35 1.1 Introduction
36 Integral membrane proteins play essential roles in numerous physiological
37 functions, such as molecular recognition, energy transduction and ion regulation. Despite
38 the experimental challenges of studying these proteins, they are critical to understand
39 since they represent more than 60% of drug targets [1], [2]. For example, G-protein
40 coupled receptors (GPCRs), a class of membrane proteins, are intensively studied using
41 computational resources since the malfunction of these receptors results in serious
42
43 disorders such as hypertension, congestive heart failure, stroke and cancer. On a similar scale, genetic disorders of ion channels result in ‘channelopathies’ such as cystic fibrosis,
44 Bartter syndrome and paralysis. Therefore, ongoing technological advances are exploited
45 to study membrane proteins, in order to improve or develop novel pharmacological drug
46 targets.
47 The availability of complete or partial genome sequences for a number of
48 organisms from a number of domains including the eubacterial, archaean and eukaryotic
49 domains now makes possible much more detailed studies of membrane protein topology.
50 Compounded by their genomic abundance, the use of computational tools in this field is
51 essential and timely. In combination with the advancement of simulation techniques, the
52 advent of structural genomics has spurred the membrane protein field to consider high-
53 throughput methods, which can help redress the disparity between soluble proteins and
54 membrane proteins. Indeed, numerous bioinformatics and proteomic analyses (e.g.[3],
55 [4], [5], [6], [7]) have been carried out to examine membrane protein architecture and
3
Drug Discovery Today: Informatics (Computational Drug Discovery)
56 even to closely analyze detailed stabilizing and mediating interactions between
57 transmembrane helices in membrane proteins.
58 Membrane proteins are, in many respects, easier to investigate computationally
59 than experimentally, due to the uniformity of their structure and interactions [8], [9]. The
60 high propensity to form secondary structures reduces the number of degrees of freedom,
61 which determine the protein’s fold, and hence lowers the complexity of predicting the
62 structures of these proteins. Computational techniques represent key methods for relating
63 the few static experimental membrane protein structures to dynamic biological systems,
64 thereby yielding maximum benefit from the limited structural and mechanistic
65 information available. The computational techniques employed in the biological arena to
66 explore membrane proteins are dizzyingly vast, thus in this review, we will focus on one
67 intensively utilized technique: MD simulations.
68
69 1.2 Membrane Protein Structure
70 Membrane proteins are essentially divided into two main classes: some contain a
71 significant portion of their mass within the interior of the membrane (intrinsic or integral
72 membrane protein (IMP)) while other proteins are only associated to the membrane
73 surface (extrinsic or peripheral proteins). For IMPs, two common structural motifs have
74
75 been observed for the transmembrane (TM) domains of membrane proteins: (1) α-helical or (2) a β-sheet topology [10]. These two folds [Figure 1] are the simplest solutions to
76 satisfying the hydrogen bonding potential of the polypeptide backbone amide groups
77 within the lipid bilayer. The majority of IMPs display α-helical transmembrane segments
4
Drug Discovery Today: Informatics (Computational Drug Discovery)
78 and can be further divided into two types: bitopic (those which traverse the lipid bilayer
79
80 with a single α-helix) and polytopic (an α-helical bundle).
Membrane proteins that are α-helical typically form well-packed bundles as, for example,
81 found in bacteriorhodopsin, photosynthetic reaction centers and cytochrome C oxidase.
82
Formation of β-sheets is seen in bacterial outer membrane proteins (e.g. OmpA [11] and
83 FecA [12]), which span the membrane as β-barrels.
88
89
90
91
92
93
94
95
96
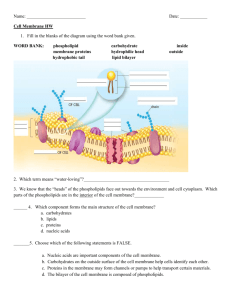
84
85
86
87
1.3 Experimental structure determination
97 The disparity between our knowledge of soluble proteins and membrane proteins
98 is largely due to the practical difficulties involved in expressing and crystallizing the
99 latter [13]. Their inherent membrane-bound nature makes structure determination a
100 particular challenge, and thus requires special treatment. This is particularly true for α-
101 helical membrane proteins as they tend to be hydrophobic and are therefore difficult to
102 unfold and refold in vitro. β-barrel proteins, on the other hand, are more hydrophilic and
103 amenable to the traditional methods of denaturation and refolding into detergents, or
104 directly into lipids [14], [15].
105 Three main bottlenecks exist with obtaining structural information of membrane
106 proteins. Firstly, it is difficult to obtain the protein of interest since membrane proteins
5
Drug Discovery Today: Informatics (Computational Drug Discovery)
107 are usually only present in the cell at low concentrations. Overexpression is therefore a
108 necessity for the majority of membrane proteins that cannot be readily obtained in
109 sufficient amounts from their native environments [16]. Many different expression
110 systems are used though each have their drawbacks, including low yield (often due to
111 toxicity), heterogeneous post-translational modification, low stability and partial
112 proteolysis [13]. The majority of membrane protein crystal structures result from proteins
113 that naturally occur at high concentrations or have been overexpressed in a homologous
114 system. Secondly, membrane proteins are naturally embedded in a heterogeneous
115 dynamic environment of the mosaic lipid bilayer [Figure 2] and it is impossible to use
116 high-resolution experimental techniques in their native environment.
117
118
119
120
121
122
123
124
125
126 The proteins therefore need to be extracted from the native membrane and studied in
127 detergent or lipid environment in vitro, which leads to difficulties in sample preparation
128 for biophysical methods, such as X-ray crystallography and NMR. However, cryo-
129 electron microscopic analysis differs from these techniques in that it can be used to study
130 membrane proteins in a crystalline or non-crystalline state at high resolution [17]. This
6
Drug Discovery Today: Informatics (Computational Drug Discovery)
131 has enabled, for example, the structure of bacteriorhodopsin to be analyzed to a resolution
132 of 2.8Å [18], [19]. Thirdly, membrane proteins are generally insoluble in aqueous
133 solution hence detergents are required in concentrations above the critical micellar
134 concentration (CMC). Too much detergent can denature the protein or impede
135 crystallization by phase separation, whilst too little and the protein may become
136 insoluble. The production of three-dimensional (3D) or two-dimensional (2D) crystals
137 remains one of the major challenges in obtaining structural information.
138 Despite the inherent difficulties in studying the structure of membrane proteins,
139 they remain a crucial area of study due to their essential role in the control of important
140 biochemical processes. A number of experimental methods exist and are continually
141 being developed with the aid of technological tools to extract structural information on
142 membrane proteins. Spectroscopic methods [20], [21] such as vibrational spectroscopy,
143 Raman, FTIR and circular dichroism (CD) have been utilized to determine their
144 secondary structure and to help distinguish between competing models of structure or
145 function. Bacteriorhodopsin [22], the acetylcholine receptor [23], lactose permease [24]
146 and the outer membrane proteins of E. coli [25] are examples of membrane proteins
147 whose secondary structure content have been determined with such techniques.
148 Alternative methods, namely crystallographic techniques, have since been used to
149 determine high-resolution structures. Three methods are generally employed: electron
150 microscopy, NMR and X-ray crystallography. Despite the difficulties involved in
151 generating large and sufficiently well-ordered 3D crystals, X-ray crystallography is still
152 the most successful and least difficult technique for obtaining high-resolution structures.
153 Electron crystallography [26] and atomic force microscopy (AFM) [27], [28] are also
7
Drug Discovery Today: Informatics (Computational Drug Discovery)
154 used as methods for membrane proteins whose natural propensity is to form 2D arrays
155 [29].
167
168
169
170
171
172
173
174
175
176
177
156 Each method has its own advantages and hence the structural data obtained are
157 complementary. All methods are generally used in parallel in an attempt to achieve the
158 best structural description of a membrane protein. In addition, methods are continually
159 being developed, with the use of computational resources, leading to an increasing rate of
160 membrane protein structure determination. From the plot [Figure 3], the growth of known
161 structures is exponential with a growth rate of ~1.3-fold per year, but lags behind the rate
162 for soluble proteins during the equivalent time period. Assuming a continuous
163 exponential rate of growth in the number of structures determined, we would expect close
164 to 300 structures to be known by the end of 2008. Despite the increasing rate of structure
165 determination, improved structure prediction methods combined with computational tools
166 are important in studying membrane proteins.
3
2.5
2
1.5
1
0.5
0
1991 1992 1993 1994 1995 1996 1997 1998 1999 2000 2001 2002 2003 2004 2005 2006 2007
Year
8
Drug Discovery Today: Informatics (Computational Drug Discovery)
178 1.4 Computational Structure Determination
179 Computer simulation methods have provided key insights into the general nature
180 of protein motion and aspects of motion linked to the function of proteins in their native
181 state. They are rapidly becoming a standard tool to study the structure and dynamics of
182 membrane proteins. With the increasing number of high-resolution structures of
183 membrane proteins, a wide range of membrane proteins can now be simulated over time
184 spans that capture essential biological processes. Whilst X-ray structures of membrane
185 proteins provide static, spatially and temporally averaged snapshots of the proteins in
186 specific crystal environments, simulations enable us to explore the structural dynamics of
187 the proteins in an attempt to bridge the gap between structure and function of proteins. By
188 employing computational methods one can combine both experimental and theoretical
189 data of ion channels in order to describe their physiological properties in terms of
190 underlying physical processes.
191 One of the main challenges is to relate molecular structures to the physiological
192 properties of the protein. For ion channels this has been addressed by describing the
193 structure of the channel at varying levels of detail and accuracy, where the key regions
194 can be broken down into the transbilayer pore, the selectivity filter and the gate. A wide
195 variety of computational approaches such as molecular dynamics (MD) simulations (e.g.
196 [30], [31], [32]), continuum electrostatic Poisson-Boltzmann (PB) theory [33], [34],
197 Brownian dynamics (BD) [35], [36], electrodiffusion theory [37] have helped to refine
198 our understanding of the molecular determinants of channel function. MD arguably
199 provides the most detailed information in the theoretical studies of membrane proteins.
9
Drug Discovery Today: Informatics (Computational Drug Discovery)
200 In an MD simulation, all atoms in the system (including ions and water
201 molecules) are represented explicitly. In the classical fine-grained approach, simulations
202 are typically carried out using empirically determined pairwise interaction potentials
203 between the atoms. Another MD approach, known as ab initio MD, uses interactions
204 between atoms determined from first principles electronic structure calculations. Since
205 there are no free parameters with this approach, it could potentially be the ideal approach
206 to modeling ion channels. However, due to the extremely demanding nature of the
207 computations, its applications are currently limited to very small systems.
208 With the atomistic approach of classical MD simulations, rapid advances in
209 simulation methodologies and computational power has led to accessible timescales of up
210 to 0.1
s. Amongst all structurally known membrane proteins, the ion channels (KvAP,
211 KcsA), transporters (AQP, GlpF, ABC transporter), and outer membrane proteins
212 (OmpA) have been examined via simulation in particular detail. The methods described
213 will be briefly compared using the bacterial K
+
channel, KcsA, as a case study. This is the
214 first biological ion channel whose tertiary structure was elucidated [38] and has been
215 studied extensively in terms of ion selectivity, permeation and gating.
216
217 1.5 Comparisons between MD techniques
218 MD simulations on KcsA have been employed to examine channel selectivity, ion
219 permeation and ion transport energetics in potassium channels, with the main focus being
220 on the selectivity filter and understanding the permeation properties of K
+
ions in the
221 filter and cavity region. Many of the results from MD simulations based on realistic all-
10
Drug Discovery Today: Informatics (Computational Drug Discovery)
222 atom models have been consistent with the information obtained from high-resolution
223 structural data [39].
224 The scope of MD simulations can be extended by using commonly utilized
225 algorithms. For example, three methods employed are (i) umbrella sampling [40]; (ii) the
226 application of external forces to the system, such as using an expanding sphere inside the
227 pore at the gate region of the KcsA channel to induce gating [41], and with the
228 application of steered-MD; (iii) alchemical free energy perturbation (FEP) [42][43].
229 Although results from these techniques increase our confidence in MD, we cannot build a
230 complete picture of ion permeation if ion fluxes cannot be simulated or if channel
231 conductance cannot be calculated. Single-channel measurements reveal the net
232 translocation of one ion in KcsA to be in 10-20 ns [44] which is the order of timescales
233 accessible by MD simulations [42][45]).
234 Despite the significant increase in computational power and molecular dynamics
235 methods, the direct simulation of ionic fluxes across channels and large conformational
236 changes using an atomistic description remains computationally challenging at present.
237 This is due to time scales of 10 – 100 ns being much shorter than the typical timescale for
238 allosteric effects which is usually in the order of microseconds to milliseconds. Thus,
239 biased MD methods, such as steered MD (e.g. [46]) and targeted MD (e.g. [47]) have
240 been developed to circumvent these time scale limitations. However, the above described
241 weakness of MD is the strength of Brownian Dynamics (BD). The drawback of BD is,
242 however, the comparatively poor description or parameterization of the biological system
243 simulated. Thus, diffusion coefficients and free energies for example, which can be
11
Drug Discovery Today: Informatics (Computational Drug Discovery)
244 determined with MD, cannot be calculated with BD. Hence, both BD and MD are
245 complementary.
246 In BD, ion permeation can be simulated for sufficiently long to measure channel
247 conductance without having to treat a system in all atomic details explicitly. BD
248 simulations (for example on KcsA [48] [49]) treat protein atoms forming the channel as
249 rigid, and the water implicitly as a dielectric continuum performing Brownian motion.
250 Despite these severe limitations of the continuum electrostatic approximation and the
251 assumption of a rigid channel structure, BD simulations [50] confirmed the multi-ion
252 mechanism to be in agreement with the ion flux determined experimentally [51]. The
253 ability to compute current flow across ion channels confers a distinct advantage to BD
254 simulations over other techniques with the applications of BD to calculating current and
255 voltage conductance in ion channels. The assumptions in BD of treating the water-protein
256 interface as a rigid boundary and the treatment of water in a narrow pore as a continuum
257 are simplifications since proteins (and lipid bilayers) are in fact dynamical, undergoing
258 fluctuations on a picosecond timescale, which is much more rapid that the timescale for
259 ion permeation.
260 The treatment of the water-protein boundary has been modified in some studies in
261 an attempt to reduce its simplified stochastic nature. For example, an elaborate treatment
262 of boundaries was proposed [52][53] using a grand canonical Monte Carlo (GCMC)
263 method. However, comparison of BD using a simple stochastic boundary and the GCMC
264 boundary [54] revealed no significant differences with the results obtained when the
265 boundaries were at a reasonable distance from the channel. MD is also the preferred
12
Drug Discovery Today: Informatics (Computational Drug Discovery)
266 technique for size-dependent selectivity among ions with the same valence, since such
267 ions cannot be distinguished in BD. Whilst microscopic quantities can be deduced from
268 BD, the increased high-level detail adopted by MD and Monte Carlo (MC) algorithms
269 enables the analysis of large-scale conformational changes. For example, one study [55]
270 explored the conformational changes between the open (KcsA crystal structure [38] and
271 closed forms (model generated from MthK [56]) of KcsA. The simulation of the large-
272 scale conformational transition was run by imposing lateral forces to the C-termini of the
273 inner helices and minimizing the energy at each step. As a result of the applied forces the
274 inner helices converged to form a tightly packed structure, with a change in backbone
275 geometry in the central region.
276 Whilst MD provides the most detailed information about the dynamics of ion
277 channels, currently accessible simulation times are its greatest limitation. Hence it is
278 unclear at present if the results obtained from MD simulations reflect reality or are an
279 artifact of the method. However, this problem seems sure to be surmounted in the future
280 with the doubling of computer speeds over the years. In the meantime faster, more
281 coarse-grained methods are being employed [57][58] to calculate the conductance of ion
282 channels. In addition, free energy methods such as replica exchange and ensemble
283 dynamics [59][60] are methods that are becoming increasingly viable with increasing
284 computational power. Despite such hurdles, MD simulations in combination with other
285 computational tools such as homology modelling, and experimental studies such as
286 mutagenesis analysis, have proved to be essential in the study of membrane protein
287 structure and function, which in turn enables the development of novel pharmacological
288 drug targets.
13
Drug Discovery Today: Informatics (Computational Drug Discovery)
289 1.6 Conclusions and Future Outlooks
290 Integral membrane proteins (IMPs) perform key functions in regulating the
291 physiological state of the cell. This is especially true for receptors and ion channels that
292 control, for example, the transmembrane (TM) potential. The scarcity of IMP structures is
293 due to the fact that the route from membrane protein sequences to atomic-resolution
294 structures is not as straightforward as for their soluble counterparts. This is, in turn,
295 primarily due to the substantial difficulties with overexpression and crystallization of
296 IMPs. Thus, the use of computational tools such as protein simulation methods, in
297 combination with experimental and structural genomic studies is becoming increasingly
298 valuable in studying the structure and function of membrane proteins.
299 The explosion of genomic data in combination with huge advances in
300 computational resources and experimental techniques is leading to a greater
301 understanding of biological structure, function and mechanisms. Considering the
302 dramatic advancements in molecular dynamics simulation methodologies in recent years,
303 it is likely that current drawbacks will be overcome considerably in the near future.
304 Reassuringly, over the last few years there has been a dramatic increase in the number of
305 membrane protein crystal structures obtained, with 30 structures being solved in 2006
306 alone.
307 Unsurprisingly, given the immense computing power and wealth of genomic and
308 structural data, recent years have seen a rise in structural genomics initiatives primarily
309 focusing on membrane proteins in an attempt to harness the synergy between the growing
310 data and technology available. Examples of such initiatives include the (1) Swiss
14
Drug Discovery Today: Informatics (Computational Drug Discovery)
311 National Center of Competence in Research (NCCR; www.structralbiology.ethz.ch
), (2)
312 Membrane Protein Network (MePNet; www.mepnet.org
), (3) European Membrane
313 Proteins (E-MeP; www.e-mep.org
), (4) Protein Wide Analysis of Membrane Proteins
314 (ProAMP; www.pst-ag.com
), (5) Biological Information Research Center, Japan (BRIC;
315 unit.aist.go.jp/birc) and the (6) Membrane Protein Structure Initiative (MPSI;
316 www.mpsi.ac.uk
). At present, the large majority of crystallized membrane proteins are
317 bacterial proteins, thus there is an urgent need to obtain structures of eukaryotic
318 membrane proteins as these could be potential drug targets. In this respect, structural
319 genomics initiatives are essential for rapidly increasing the structure determination
320 throughput of eukaryotic membrane proteins. Interestingly, this situation is analogous to
321 that of soluble proteins; slow structure determination in the 1970s was followed by an
322 exponential increase of structures generated due to improved experimental protocols.
323 The paradox posed by the sheer number of potential helical membrane proteins
324 and the lack of high-resolution structural and thermodynamic information for them
325 emphasizes the extensive work that remains to be done in the field of membrane proteins.
326 The potential payoff may be great as this class of proteins has historically contained
327 excellent targets for therapeutics. Advances in our ability to understand and manipulate
328 membrane proteins may lead to the discovery or design or pharmaceutical agents that can
329 modulate their functions.
330
15
Drug Discovery Today: Informatics (Computational Drug Discovery)
331 References
332
333
334
335
336
337
[1] Davey, J. (2004) G-Protein-Coupled-Receptors: New Approaches to Maximize the
Impact of GPCRs in Drug Discovery. Expert Opinion on Therapeutic Targets 8,
165-170.
[2] Terstappen, G. C., and Reggiani, A. (2001) In silico research in drug discovery.
Trends Pharmacol. Sci.
22, 23-26.
338
339
[3] Xia, Y. et al.
(2006) Integrated prediction of the helical membrane protein interactome in yeast. J. Mol. Biol. 357, 339-349.
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
340
341
342
343
344
345
346
347
348
349
350
[4] Hurwitz, N. et al.
(2006) Towards genome-scale structure prediction for transmembrane proteins. Phil. Trans. Roy. Soc. ser. B 361, 465-475.
[5] Lundstrom, K. (2005) Structural Genomics of GPCRs. Trends Biotech. 23, 103-108.
[6] Walian, P. et al. (2004) Structural Genomics of Membrane Proteins. (2004)
Genome Biol. 5, 215.
[7] Lehnert, U. et al.
(2004) Computational analysis of membrane proteins: genomic occurrence, structure prediction and helix interactions. Q. Rev. Biophys.
37, 121-
146.
[8] Sachs, J.N. and Engelman, D.M. (2006) Introduction to Membrane Protein
Reviews: Interplay between Structure, Dynamics, and Environment in Membrane
Protein Function. Annu. Rev. Biochem. 75, 707-712.
[9] Engelman, D.M. et al.
(2003) Membrane Protein Folding: Beyond the Two Stage
Model. (2003) Febbs Lett. 555, 122-125.
[10] Cowan, S. W., and Rosenbusch, J. P. (1994) Folding pattern diversity of integral membrane-proteins. Science 264, 914-916.
[11] Pautsch, A., and Schulz, G. E. (1998) Structure of the outer membrane protein A transmembrane domain. Nature Struct. Biol.
5, 1013-1017.
[12] Ferguson, A. D.
et al.
(2002) Structural basis of gating by the outer membrane transporter FecA. Science 295, 1715-1719.
[13] Grisshammer, R., and Tate, C. G. (1995) Overexpression of integral membraneproteins for structural studies. Q. Rev. Biophys.
28, 315-422.
[14] Buchanan, S. K. (1999) ß-Barrel proteins from bacterial outer membranes: structure, function and refolding. Curr. Opin. Struct. Biol.
9, 455-461.
16
Drug Discovery Today: Informatics (Computational Drug Discovery)
373
381
382
383
384
385
386
387
374
375
376
377
378
379
380
388
389
390
391
392
393
394
395
396
397
398
399
400
401
408
409
410
411
412
413
414
402
403
404
405
406
407
415
416
417
418
[15] Schulz, G. E. (2000)
-Barrel membrane proteins. Curr. Opin. Struct. Biol.
10, 443-
447.
[16] Tate, C. G. (2001) Overexpression of mammalian integral membrane proteins for structural studies. FEBS Lett.
504, 94-98.
[17] Chiu, W. (1993) What does electron cryomicroscopy provide that X-ray crystallography and NMR spectroscopy cannot? Ann. Rev. Biophys. Biomol. Struct.
22, 233-355.
[18] Baldwin, J. M., Henderson, R., Beckmann, E., & Zemlin, F. (1998) Images of purple membrane at 2.8Å resolution obtained by cryo-electron microscopy.
J. Mol.
Biol.
202, 585-591.
[19] Fujiyoshi, Y. (1998) The structural study of membrane proteins by electron crystallography. Adv. Biophys.
35, 25-80.
[20] Bazzi, M. and Woody, R. W. (1985) Oriented secondary structure in integral membrane proteins. Biophys. J.
48, 957-966.
[21] Krimm, S. and Bandekar, J. (1986) Vibrational spectroscopy and conformation of peptides, polypeptides and proteins. Adv. Protein Chem.
38, 183-364.
[22] Glaeser, R. M. et al . (1991) What spectroscopy can still tell us about the secondary structure of bacteriorhodopsin. Biophys. J.
59, 934-938.
[24] Yager, P. et al. (1984) The secondary structure of acetylcholine receptor reconstituted in a single lipid component as determined by Raman spectroscopy.
Biophys. J.
45, 26-28.
[25] Vogel, H. et al.
(1985) The structure of the lactose permease derived from Raman spectroscopy and prediction methods. EMBO J.
4, 3625-3631.
[25] Vogel, H. and Jähnig, E. (1986) Models for the structure of outer-membrane proteins of Escherichia coli derived from raman spectroscopy and prediction methods. J. Mol. Biol. 190, 191-199.
[26] Saibil, H. R. (2000) Conformational changes studied by cryo-electron microscopy.
Nature 7, 711-717.
[27] Clausen-Schaumann, et al.
(2000) Force spectroscopy with single bio-molecules.
Curr. Opin. Chem. Biol.
4, 524-530.
[28] Scheuring, S. et al. (2002) Sampling the conformational space of membrane protein surfaces with the AFM. Eur. Biophys. J.
31, 172-178.
17
Drug Discovery Today: Informatics (Computational Drug Discovery)
419
420
[29] Stahlberg, H. et al.
(2001) Two-dimensional crystals: a powerful approach to assess structure, function and dynamics of membrane proteins. FEBS Lett.
504, 166-172.
421
422
423
[30] Sotomayor, M., Vasquez, V., Perozo, E. & Schulten, K. (2007) Ion Conduction through MscS as Determined by Electrophysiology and Simulation. Biophys. J. 92,
886-902.
424
425
[31] Saparov, S.M. et al.
(2007) Fast and Selective Ammonia Transport by Aquaporin-8.
J. Biol. Chem. 282, 5296-5301.
426
427
[32] Mukherjee, P. et al.
(2006) Picosecond dynamics of a membrane protein revealed by 2D IR. Proc. Natl. Acad. Sci. U.S.A. 103, 3528-3533.
428
429
430
[33] Peter, C. and Hummer, G. (2005) Ion Transport through Membrane-Spanning
Nanopores Studied by Molecular Dynamics Simulations and Continuum
Electrostatics Calculations Biophys. J.
89, 2222-2234.
431
432
[34] Sengupta, D. et al. (2005) Understanding the energetics of helical peptide orientation in membranes. Proteins 58, 913-922.
440
441
442
443
444
445
446
447
448
449
450
451
452
453
433
434
435
436
437
438
439
454
455
456
457
458
[35] Sieber, J.J. et al.
(2007) Anatomy and Dynamics of a Supramolecular Membrane
Protein Cluster. Science 317, 1072-1076
[36] Jin, S. et al.
(2007) Single-Particle Tracking of Membrane Protein Diffusion in a
Potential: Simulation, Detection, and Application to Confined Diffusion of CFTR
Cl
–
Channels. Biophys. J. 93, 1079-1088.
[37] Coalson, R.D. (2005) Poisson-Nernst-Plank Theory approach to the calculation of current through biological ion channels. Nanobiosci.
4, 81-93.
[38] Doyle, D. A. et al.
(1998) The structure of the potassium channel: molecular basis of K
+
conduction and selectivity. Science 280, 69-77.
[39] Roux, B. (2002) Theoretical and computational models of ion channels. Curr. Opin.
Struct. Biol.
12, 182-189.
[40] Crouzy, S. et al.
(2001) Extracellular blockade of K
+
channels by TEA: Results from molecular dynamics simulations of the KcsA channel. J. Gen. Physiol.
118,
207-217.
[41] Biggin, P. C. and Sansom, M. S. P. (2002) Open-state models of a potassium channel. Biophys. J.
83, 1867-1876.
[42] Åqvist, J. and Luzhkov, V. (2000) Ion permeation mechanism of the potassium channel. Nature 404, 881-884.
18
Drug Discovery Today: Informatics (Computational Drug Discovery)
480
481
482
483
484
485
486
487
488
489
490
491
492
493
473
474
475
476
477
478
479
494
495
496
497
498
499
500
501
502
503
504
459
460
461
462
463
464
465
466
467
468
469
470
471
472
[43] Berneche, S. and Roux, B. (2002) The Ionization State and the Conformation of
Glu-71 in the KcsA Channel. Biophys. J. 82, 772-780.
[44] LeMasurier, M. et al. (2001) KcsA: It's a potassium channel. J. Gen. Physiol.
118,
303-313.
[45] Domene, C. and Sansom, M. S. P. (2003) Potassium channel, ions and water: simulation studies based on the high-resolution X-ray structure of KcsA. Biophys. J.
85, 2787-2800.
[46] Treptow, W. and Tarek, M. (2006) K
+
conduction in the selectivity filter of potassium channels is monitored by the charge distribution along their sequence.
Biochem. 91, L81-L83.
[47] Compoint, P. et al.
(2006) Targeted Molecular Dynamics of an open-state KcsA channel. J. Chem. Phys. 13, 134707.
[48] Allen, T. W. and Chung, S. H. (2001) Brownian dynamics study of an open-state
KcsA potassium channel. Biochim. Biophys. Acta . 1515, 83-91.
[49] Burykin, A. et al.
(2003) Exploring the origin of the ion selectivity of the KcsA potassium channel. Proteins: Structure Function and Genetics 52, 412-426.
[50] Chung, S. H. et al. (2002) Conducting-state properties of the KcsA potassium channel from molecular and Brownian dynamics simulations. Biophys. J.
82, 628-
645.
[51] Ermak, D. L. and McCammon, J. A. (1978) Brownian dynamics with hydrodynamic interactions. J. Chem. Phys.
, 69, 1352-1360.
[52] Im, W. et al (2000) A Grand Canonical Monte Carlo-Brownian dynamics algorithm for simulating ion channels. Biophys. J.
79, 788-801.
[53] Woo, H.-J. et al.
(2004) Grand canonical Monte Carlo simulations of water in protein environments. J. Chem. Phys.
121, 6392-6982.
[54] Corry, B. et al.
(2002) Reservoir Boundaries in Brownian Dynamics Simulations of
Ion Channels. Biophys. J.
82, 1975-1984.
[55] Tikhonov, D. B. and Zhorov, B. S. (2004) In Silico Activation of KcsA K + channel by Lateral Forces Applied to the C-termini of Inner Helices. Biophys. J.
87, 1526-
1536.
[56] Jiang, Y., et al.
(2002) The open pore conformation of potassium channels. Nature
417, 523-526.
19
Drug Discovery Today: Informatics (Computational Drug Discovery)
505
506
507
508
509
510
511
512
513
514
515
516
517
[57] Brannigan, G. et al.
(2007) Implicit solvent simulation models for biomembranes.
Eur. Biophys. J. 35, 104-124.
[58] Lei, H. and Duan, Y. (2007) Improved sampling methods for molecular simulation.
Curr. Opin. Struct. Biol. 17, 187-191.
[59] Bu, L. et al. Membrane Assembly of Simple Helix Homo-Oligomers studied via molecular dynamics simulations. Biophys. J. 92, 854-863.
[60] Chen, Z. and Xu, Y. (2006) Energetics and stability of transmembrane helix packing: A replica-exchange simulation with a knowledge-based membrane potential. Proteins: Structure Function and Genetics 62, 539-552
20
Drug Discovery Today: Informatics (Computational Drug Discovery)
518
519
520
Figure Legends
Figure 1: Two examples of membrane proteins. KcsA (left) (PDB: 1K4C) is a voltage-
521 gated K+ selective α-helical protein. OmpA (right) (PDB: 1QJP) is an example of a β-
522 barrel membrane protein. The dashed lines indicate the position of the bilayer.
523
524
525 Figure 2: Illustration of a membrane protein (KcsA, shown in purple) embedded in a lipid
526 bilayer. For clarity, the water molecules on either side of the lipid bilayer have not been
527
528 included. The hydrocarbon core of a membrane is typically ~25-30Å wide with the headgroups spanning ~10Å. The polar head groups of the lipids face the aqueous
529 environment on both sides of the membrane, whereas their hydrophobic chains form the
530 insulating interior of the bilayer. Owing to the ester carbonyls and water associated to the
531 lipid headgroups, lipid molecules possess electrical dipoles which result in a considerable
532 electrical potential (positive inside the bilayer). (Figure generated using KcsA crystal
533 structure, PDB: 1K4C).
534
535
536 Figure 3: Rate at which new structures (α-helical and β-barrel) have been determined
537 since 1991. The count of membrane protein structures includes the same protein from
538 different organisms. The data suggests that there will be ~300 structures at the end of
539 2008. Data extracted from http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html
540
21