EPAPS_broeker_corrected
advertisement

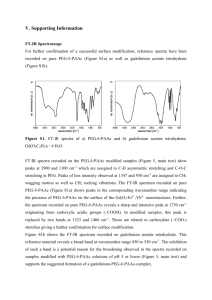
EPAPS Document for Density dependent reorientation and rehybridization of chemisorbed conjugated molecules for controlling interface electronic structure B. Bröker1, O. T. Hofmann2, G.M. Rangger2, P. Frank2, R. P. Blum1, R. Rieger3, L. Venema4, A. Vollmer5, K. Müllen3, J. P. Rabe1, A. Winkler2, P. Rudolf4, E. Zojer2, and N. Koch1,* 1 Institut für Physik, Humboldt-Universität zu Berlin, Newtonstrasse 15, D-12389 Berlin, Germany. 2 Institute of Solid State Physics, Graz University of Technology, Petersgasse 16, A-8010 Graz, Austria. 3 Max Planck Institut für Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany. 4 Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. 5 Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, D-12489 Berlin, Germany. *email: nkoch@physik.hu-berlin.de 1 1. Additional methodological details Experimental: UPS experiments were performed at the endstation SurICat (beamline PM4) at the synchrotron light source BESSY II (Berlin, Germany) [1]. Spectra were collected in normal emission with a hemispherical electron energy analyzer (Scienta SES 100) using an excitation photon energy of 35 eV and an overall energy resolution of 150 meV. RAIRS experiments were conducted at the Zernike Institute of Advanced Materials using a Bruker IFS 66v/S spectrometer. The IR beam entered the UHV system through a KBr window and was directed onto the sample under an angle of 83° (to the surface normal). For detection the reflected beam left the chamber through another KBr window and hit a LN2 cooled HgCdTe detector. The spectrometer and the beam path from and to the UHV chamber were evacuated and the pressure kept in the mbar range. The spectra were recorded from 500 to 3000 cm-1, the resolution of the spectrometer was set to 4 cm-1 and typically 500 scans were co-added which gives rise to a total acquisition time of about 3 minutes per spectrum. The spectrum of the clean substrate was taken as a background reference R0 at the beginning of each experiment. Subsequent spectra were recorded in a time evolved mode during HATCN deposition and are displayed in reflectance as the ratio of the difference to the background spectrum (R-R0)/R0. The coverages listed in the figures are the nominal thicknesses read from the quartz crystal microbalance (QCM) before a new spectrum was acquired. TDS was carried out using a quadrupole mass spectrometer (QMS). The QMS was tuned to the most relevant masses which were identified in advance by desorbing HATCN directly from the source into the QMS. The substrate could be heated up to 900 K by resistive heating. The same setup was also equipped with an UHV compatible Kelvin probe (KP) allowing for work function measurements. Advanced “off-null” detection as well as automatic control of the tip-tosample spacing was used, which is an important requirement for accurate measurements [2]. 2 All three experimental setups consist of interconnected sample preparation (base pressure < 5x10-8 mbar) and analysis (base pressure 1x10-10 mbar) chambers, which enable sample transfer without breaking vacuum. The Ag(111) single crystals were cleaned by repeated cycles of annealing (up to 550 °C) and Ar-ion sputtering. HATCN was synthesized according to the literature procedure [3] and sublimed from resistively heated tantalum pinhole sources. Prior to the experiments the HATCN evaporators were thoroughly out-gassed for at least 2 days. The mass-thickness of the organic layers (i.e., the coverage used throughout the manuscript) was monitored with a quartz crystal microbalance (QCM) in all experimental setups, and a HATCN density of 1.6 g/cm3 from Ref. [4] was used. The same sticking coefficient was assumed on both the quartz crystal of the QCMs and the Ag(111) crystals. The values obtained from the QCMs in the various experiments all coincided within 10% for the transitions between regions I-III, thus providing good confidence in the reported values. Theoretical description: Density functional theory calculations were performed using the 3D periodic VASP code [5, 6]. The PW91 functional was employed along with the cutoff for the plane wave basis set of 20 Ryd. For surface calculations, the periodicity in z-direction was broken by inserting a sufficiently large amount of vacuum between the slabs (>20 Å). In the vacuum region, a dipole layer was inserted to prevent artificial polarization of the periodic replicas. For the face-on-structure, more details of the methodology are reported elsewhere [7]. The edge-on structure was computed in a 2x3√3 Ag (111) unit cell, using 15x6x1 k-points. All other details are equivalent to the face-on calculations. For the sake of consistency, gas phase calculations were also performed in VASP, using the same unit cell as for the periodic structure. When simulating isolated molecule, dipole layers were introduced in the surrounding vacuum in all 3 spatial directions. 3 For the upright standing conformation, an orthogonal, 2x3√3 unit cell was assumed. This geometry corresponds to π-π-stacking distances between the individual HATCN cores of 5.88Å and a lateral distance of 15.28Å between the centers of neighboring molecules. Upright standing HATCN supposedly adsorbs with two CN-groups pointing directly towards the surface. There are two different possibilities to realize this situation. Either, the molecule could adsorb on its “cusp”, i.e. with adjacent cyano groups. Alternatively, the molecule could adsorb 60° rotated to the former structure, and two cyano groups of different rings point toward the surface. Both conformations are shown in Fig. S1. Electronically, both geometries yield similar results. The work-function modification is computed to be +2.48 eV for the “cusp”-geometry and +2.37 eV for the 60° rotated one. Slight geometrical distortions can be observed in both conformations. These are attributed to the incommensurability between the CN-group spacing and the spacing between the optimal docking site. In neither geometry is a molecular dipole induced (µMol < 0.1 eV). All work-function modifications must therefore be attributed to charge-transfer processes. The methodology applied here has been tested, for example, for the experimentally presumably best characterized metal / organic semiconductor interfaces, namely the interfaces between PTCDA and noble metals [8]. Romaner et al. have shown that the calculated workfunction modifications induced by a 3,4,9,10-perylene-tetracarboxylicdianhydride PTCDA monolayer on Cu(111), Ag(111) and Au(111) surfaces that results primarily from interfacial charge transfer is in excellent agreement with experimental data as long as the adsorption geometry is properly described [9]. They showed that for the correct adsorption distance, methodological details like the chosen exchange-correlation functional 4 play an only very minor role; an inaccurately determined adsorption geometry, however, will result in inaccurate predictions of work-function changes. This can become a serious complication, as van der Waals interactions are not properly accounted for by (semi)local functionals and in weakly bonded systems even non-local functionals provide results that only qualitatively but not quantitatively agree with experiments [9]. For flat-lying HATCN this issue is significantly mediated by the interaction of the –CN groups with the surface as discussed for the related molecule 2,3,5,6-tetrafuoro7,7,8,8-tetracyano-quinodimethane (F4TCNQ) on Cu(111) [10] and Ag(111) [11]. In [10] a comparison between (semi) local DFT calculations and X-ray standing wave (XSW) experiments showed a good agreement for the adsorption geometry. Not surprisingly, in [10] and [11] also the calculated work-function changes were in very good accord with the corresponding experiments. In the optimization of the HATCN structure on Ag(111), we find a vertical distance between topmost metal layer and molecule varying between 2.54 Å (lowest cyano nitrogen atom) and 3.65 Å (topmost, inner ring carbon atom), which is in very close agreement with the results for F4TCNQ on Ag(111) (Ag - ring distance of 3.61 Å) [11]. These aspects lend credibility to the calculated structure of lying HATCN on Ag(111), even if the binding energy is very likely strongly underestimated. The latter forbids using the results of the DFT calculations for a quantitative analysis of the driving forces for the structural rearrangements, but in no way affects the reported work-function changes. As far as the edge-on structure is concerned, even more profound charge rearrangements occur between the downward pointing CN groups and the Ag(111) surface, i.e., also here van der Waals interactions are only one of the contributions to the actual bonding. In this context, 5 it needs to be stressed that for that conformation our calculations only serve as an estimate of how large work-function modifications could possibly be expected in an edge-on layer. This is because there is no information, whatsoever, on the actual geometry of the edge-on monolayer (see also main text) in contrast to the flat-lying layer for which STM data exist [7]. The main impact of inter-molecular interactions on the structure of an upright-standing layer can be gauged from recent combined experimental and theoretical investigations of a covalently bonded SAM (anthraceneselenolate) on Au(111) [12]. The data in this paper indicate that the molecular tilt relative to the surface normal is the most strongly affected quantity. Considering, however, that our goal here is to set an upper limit to the work-function changes in the edge-on monolayer, the study of a virtually upright standing layer (cf. Figure S1) is certainly advisable. In this context, it should also be mentioned that also in [12] an excellent agreement between measured and calculated work-function changes is found. 6 Figure S1. Front view (top) top view (middle) and side view (bottom) of upright standing HATCN adsorbing with its cusp (right) and 60° rotated (left). The unit cell is reproduced 6 times in the x and 3 times in the y direction. 7 2. Further experimental spectra and fits Vibrational mode assignment for the C≡N region The (isolated) HATCN molecule has six C≡N vibrations, of which two are pairs of twofold degenerate E’ modes and the remaining two are of A1’ and A2’ character (D3h symmetry). All modes are confined to a very narrow energy range (calculated to be within 10 cm-1). In the C≡N region of the IR transmission spectrum of neutral HATCN in KBr two pairs of degenerate E’ modes are visible, a strong one at 2241 cm-1 and a weaker one at 2251 cm-1 (Fig. 3). In the RAIRS spectra in Fig. 3, the broad mode at 2185 cm-1 is derived from the totally symmetric mode A1’, which is IR active, because of the dynamical charge transfer between molecule and surface caused by the vibration (see fit below) and a bent non-planar conformation of the molecule as stated in the main text. Mode II at 2229 cm-1 is attributed to symmetric (and thus IR active) stretching vibration of the bonded C≡N groups in regime (ii). Mode III is very sharp and its position is close to the E’ modes of HATCN in KBr, which make it reasonable to assume that this mode has also E’ character. Fitting of the RAIRS spectra from regime (i) The dotted spectrum in Fig. S2 shows the experimental RAIRS spectrum of Fig. 3, which was collected from a HATCN film with a nominal thickness of 1.8 Å. The red line represents a fit to the experimental data using a Fano lineshape from Ref. [13, 14]: 2 2 2 1 ( r ) L( ) cr 2 2 2 2 () ( r ) (1) with c being a constant used to adjust L to the experimental data, the full-width at halfmaximum of the line shape, the tunneling rate and r is the fully renormalized vibrational frequency. 8 Figure S2. Fit using a Fano-type lineshape to the data of the RAIRS spectrum of Fig. 3b, which was collected from a HATCN film with a nominal thickness of 1.8 Å. The fitting parameters are = 43 cm-1, = 0.000123 cm and r =2185 cm-1. Spectra region of the in-plane vibrational modes as a function of coverage RAIR spectra of the region from 1000 to 1800 cm-1 are shown in Fig. S3 for film thicknesses ranging from 0 to 15.4 Å. The colours of the different modes in the spectra have been chosen to fit with the assigned regimes [i.e. (i) red, (ii) black and (iii) green] as described in the main text. At the coverage corresponding to a nominal thickness of 1.8 Å coverage two broad modes can clearly be observed at ~1249 cm-1 and ~1384 cm-1 (shown as the red spectra in Fig. S3a and b). They possibly belong to totally symmetric modes, that become IR-active due to the interaction (conformational change as shown in Fig. 4 and possibly dynamical charge transfer) of face-on HATCN with the Ag(111) substrate [15]. At the onset of regime (ii), both modes start to vanish and another mode at 1331 cm-1 appears (marked black in the spectra corresponding to 2.7 Å and 3.8 Å, shown in Fig. S3a and b). This mode is attributed to the molecules starting to assume the edge-on configuration in regime (ii). With increasing 9 coverage it becomes hidden under the strong modes of the neutral molecule. The decrease of the two modes of regime (i) indicates that the face-on molecules are starting to re-orient and become incorporated into an edge-on molecular monolayer. With increasing HATCN deposition all modes of the neutral molecule [4] appear in the spectrum (green spectra in Fig. S3a and b compared to the blue HATCN KBr spectrum). The strongest ones are located at 1146, 1227 and 1342 cm-1 and are assigned to in-plane breathing modes of the HAT-backbone by DFT gas phase calculations. They can only be observed if the molecule is in an upright conformation and are thus attributed to regime (iii). Figure S3. Reflection absorption infrared spectra (RAIS) recorded during HATCN deposition on Ag(111) for the region between 1000 and 1800 cm-1. Also shown is the spectrum of HATCN pressed in a KBr disc and DFT calculated modes for the neutral molecule. Note that the KBr and the calculated spectrum have been scaled to fit the plot, besides the x-scale of the latter has been divided by 1.025. 10 UPS valence band spectra The reason why the increase sets in only in regime (ii) can be understood based on charge rearrangement and molecular distortion related arguments as described in the main text. From an energy level alignment point of view it appears, however, much less obvious at first sight. The experimental UPS spectra in Fig. S4 have been recorded during the sequential deposition of HATCN on Ag(111). A new intensity appears in the energy gap of the neutral molecule close to the Fermi-level (marked blue), which is ascribed to a hybrid metal-molecule state derived from the LUMO level of neutral HATCN. No obvious differences in the shape can be found for HATCN adsorbed face-on or edge-on, which is further supported by DFT calculations shown in the upper part of Fig. S4. Here, the LUMO level of the bonded HATCN molecules becomes partially filled also in both conformations, and the metal Fermi-level is pinned at the LUMO. As a result, the shift of the potential landscapes in the metal and the molecular layer and the modifications in both cases have to correspond to the difference between the electron affinity of the adsorbate (i.e., the corresponding LUMO energies) and of the bare substrate. Thus, one might come to believe that ΔΦ should be (almost) independent of the adsorption geometry. This line of arguments, however, misses an essential property of molecular layers described by Duhm et al. [16], namely that also in molecular systems the electron affinity is a quantity that depends on the direction in which an electron leaves an oriented layer. Indeed, we find the electron affinity perpendicular to the sample surface for face-on and edge-on planar HATCN molecules to differ by 1.15 eV. The difference in is further increased by the distortion-induced dipole of the adsorbed face-on molecules. 11 Figure S4. The valence band electronic structure of the sequential deposition of HATCN on Ag(111)as obtained by UPS is shown. Furthermore, the DFT computed densities of states projected onto the HATCN layer are plotted. The color coding of the spectra reflects their affiliation to the different regimes mentioned in the main text: red = regime (i), black = regime (ii), and green = regime (iii). The blue area represents the part of the metal-molecule hybrid state (derived from the former LUMO of neutral HATCN) that is partially filled upon adsorption for both conformations. EF denotes the Fermi level. 12 3. Aspects of application relevance As noted in the main manuscript, this the high achieved for standing HATCN on Ag(111) renders this system an interesting candidate as hole injecting electrode in electronic devices (e.g., OLED, OFET), as the absolute value of 5.4 eV parallels that of atomically clean Au, however, the electron "push-back" effect, which is detrimental for achieving low hole injection barriers on clean metal surfaces [17, 18] is expected to be minimized by the surface termination with the molecule. However, the present experiments were conducted in ultrahigh vacuum with single crystal metal surfaces. These conditions differ significantly from those prevailing during actual device fabrication. However, we are convinced that proper processengineering can yield the desired application relevance, including even solution-based processing. A related development was reported for the acceptor tetrafluoro- tetracyanoquinodimethane (F4TCNQ). In [10], the fundamental interaction (and bi-directional charge transfer) of F4TCNQ with a Cu(111) single crystal surface in ultrahigh vacuum was reported. In a later follow-up work [19] it was shown that the conceptual work could be extended to real-world fabrication processes, i.e., purely solution-based F4TCNQ deposition and with polycrystalline Cu electrodes. We anticipate that a similar development is possible with HATCN, and we note that the work function change obtained with HATCN surpasses that achievable with F4TCNQ (in UHV). 13 References [1] A. Vollmer et al., Eur. Phys. J. E 17, 339 (2005). [2] I. D. Baikie and P. J. Estrup, Rev. Sci. Instrum. 69, 3902 (1998). [3] J. Rademacher, K. Kanakarajan, and A. Czarnik, Synthesis(Stuttgart) , 378 (1994). [4] P. S. Szalay, J. R. Galán-Mascarós, R. Clérac, and K. R. Dunbar, Synth. Met. 122, 535 (2001). [5] G. Kresse and J. Hafner, Phys. Rev. B 48, 13115 (1993). [6] G. Kresse and D. Joubert, Physical Review B 59, 1758 (1999). [7] H. Glowatzki et al., Nano Lett. 8, 3825 (2008). [8] F. Tautz, Prog. Surf. Sci. 82, 479 (2007). [9] L. Romaner, D. Nabok, P. Puschnig, E. Zojer, and C. Ambrosch-Draxl, New J. Phys. 11, 053010 (2009). [10] L. Romaner et al., Phys. Rev. Lett. 99, 256801 (2007). [11] G. M. Rangger et al., Phys. Rev. B 79, 165306 (2009). [12] A. Track et al., J. Phys. Chem. C , 151. [13] D. C. Langreth, Phys. Rev. Lett. 54, 126 (1985). [14] J. Kröger, Rep. Prog. Phys. 69, 899 (2006). [15] F. S. Tautz et al., Physical Review B 65, 125405 (2002). [16] S. Duhm et al., Nature Mater. 7, 326 (2008). [17] P. S. Bagus, K. Hermann, and C. Wöll, J. Chem. Phys. 123, 184109 (2005). [18] H. Ishii, K. Sugiyama, E. Ito, and K. Seki, Adv. Mater. 11, 605 (1999). [19] J. Frisch, H. Glowatzki, S. Janietz, and N. Koch, Org. Electron. 10, 1459 (2009). 14