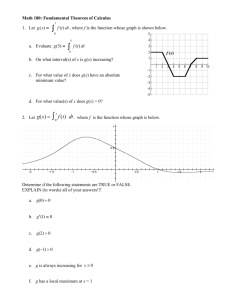
3. Actions taken in the reporting interval for - Paul-Ehrlich
advertisement

Template zur Erstellung eines: Periodic Safety Update Reports (PSUR) für ATMPs Advanced Therapy Medicinal Products Version: 001 26.06.2015 Product name Company: Street: City: Country: Date: xx.xx.2015 Paul Ehrlich Institut Paul-Ehrlich-Str. 51-59 63225 Langen Germany Periodic safety update report (PSUR) For Advanced Therapy Medicinal Products (ATMP) (Genehmigungsverfahren nach §4b AMG) Product: Active Substance: MAH: Authorisation status: Authorisation number: national PEI.A.XXXXX.01.1 Date of authorisation in Germany International Birthdate XX.XX.XXXX XX.XX.XXXX Reporting period: XX-XXX-201X to XX-XXX-201X Date of report: XX-XXX-201X Contact person for the procedure: Name: Contact details: Signature: ______________________________________ Product name Please read before starting!!! This template is intended to provide guidance for preparation of the PSUR document. It needs to be tailored to the specific product that is subject of the specific PSUR. For example: If the tabular form does not suit your needs just fill in the chapter in an appropriate, suitable form or modify the tables according to your requirements. Most likely some of the questions may not concern your product or in certain settings data required are still missing due to ongoing studies. Please state that these data are missing / lacking or write “not applicable” but do not delete the concerned section. Grey marked sections give you further advice or examples regarding the topics that should be discussed in the respective sections. Please Note: The template only covers the requirements for a PSUR in general. It thus should only be used for PSURs which concern nationally authorized ATMPs in Germany. Product name Table of contents List of Abbreviations: ...................................................................................................................................... 5 1. Introduction ............................................................................................................................................... 6 Product details ................................................................................................................................................... 6 2. Worldwide marketing authorisation Status.................................................................................... 6 3. Actions taken in the reporting interval for safety reasons ...................................................... 6 3.1. Actions related to investigational uses ....................................................................................... 6 3.2. Actions related to marketing experience ................................................................................... 6 3.3. Regulatory actions taken for safety reasons by Authorities ............................................... 6 4. Changes to Reference Safety Information ..................................................................................... 6 5.1. Exposure in Clinical Studies ............................................................................................................ 7 5.2. Cumulative and interval patient exposure from marketing experience ......................... 7 6. Data in Summary Tabulations ............................................................................................................ 8 6.1. Reference information ....................................................................................................................... 8 6.2. Cumulative summary tabulations of serious adverse events from clinical studies .... 8 6.3. Cumulative and interval summary tabulations from post-marketing data sources... 8 7. Summary of significant findings in the reporting period .......................................................... 8 7.1. Findings from completed clinical trials ........................................................................................ 8 7.2. Findings from ongoing clinical trials ............................................................................................. 8 7.3. Findings from non-interventional studies .................................................................................. 9 7.4. Findings from long-term follow-up ............................................................................................... 9 7.5. Findings from literature or independent studies ..................................................................... 9 7.6. Findings from non-clinical studies................................................................................................. 9 9. Literature .................................................................................................................................................... 9 10. Lack of efficacy in controlled clinical trials................................................................................. 9 11. Late-Breaking Information ............................................................................................................... 9 11.1. Risk minimisation measures/other actions ........................................................................... 9 11.2. Ongoing risk minimisation measures ...................................................................................... 9 11.3. Newly initiated risk minimisation measures (reporting period) .................................... 9 11.4. Planned risk minimisation measures ..................................................................................... 10 12. Other Information ............................................................................................................................. 10 13. Signal evaluation and detection .................................................................................................. 10 13.1. Signal detection ............................................................................................................................. 10 13.2. Closed signals ................................................................................................................................. 10 13.3. Ongoing signals ............................................................................................................................. 10 13.4. New signals ..................................................................................................................................... 10 14. Risk evaluation ................................................................................................................................... 10 14.1. Summary of Safety Concerns .................................................................................................. 10 14.2. Characterisation and evaluation of risks .............................................................................. 10 14.3. Characterisation of important potential problems with the treatment and with certain populations ........................................................................................................................................ 11 15. Benefit/Risk Assessment ................................................................................................................ 11 16. Overall Conclusions .......................................................................................................................... 11 RMS Product name List of Abbreviations: AEs Adverse Events Product name 1. Introduction This Periodic Safety Update Report (PSUR) No. X for XXXXX covers the period from 01XXX-201X to 31-XXX-201X. It is based on all available cumulative data since the international birth date (XX.XX.XXXX) and is focused on new information which has emerged since 01-XX-201X. Product details Invented name of the medicinal product (product short name) Active substance(s) (INN or common name) Pharmaco-therapeutic group (ATC Code): Brief description of product (chemical class, origin, production/modification, mode of action etc.) Indication/s (target population) Dosage Pharmaceutical form and strength (concentration) Rout of administration (See also SmPC product information). 2. Worldwide marketing authorisation Status Please state in which countries your product has a marketing authorisation and since when 3. Actions taken in the reporting interval for safety reasons 3.1. Actions related to investigational uses Please give an overview of planned and ongoing studies 3.2. Actions related to marketing experience Please provide an overview on actions performed in the reporting period e.g. quarantine measures because of contaminations… 3.3. Regulatory actions taken for safety reasons by Authorities 4. Changes to Reference Safety Information e.g.: In the beginning of the reporting interval the applicable reference safety information was the Company Core Data Sheet (CCDS) version no. X, dated XX-XXX-20XX. This version was updated on XX-XXX-20XX (version no. X) and the CCDS version no. X is now considered the reference safety information during the reporting period according to its date of effectiveness. The following changes were made in detail: • XXXXX • XXXXX Product name 5. Estimated exposure and use patterns In the interval XXX post-authorisation studies were ongoing and XXX were completed. 5.1. Exposure in Clinical Studies Cumulatively, XXX individual patients were exposed to “Product” in clinical trials. Study Number [Ref.] Country Indication Objectives No. of Exposure patients (no. of treated treatments) Status (ongoing, finished) Interventional Non-interventional If placebo treated or comparator patients were also studied in the trials please also present their relevant data and numbers. 5.2. Cumulative and interval patient exposure from marketing experience Country Number of treatments period cumulative period cumulative Describe the method(s) used to estimate the patient exposure? If feasible/possible also calculate patient-years of treatment. Patients treated Product name 6. Data in Summary Tabulations 6.1. Reference information Please state which MedDRA version was used for coding of adverse events/reactions 6.2. Cumulative summary tabulations of serious adverse events from clinical studies e.g.: Appendix XXX provides a cumulative summary tabulation of related and unrelated SAEs presented per SOC and Preferred term (PT). Please add the mentioned table in the Annex 6.3. Cumulative and interval summary tabulations from post-marketing data sources e.g.: Cumulatively, the MAH received XXX spontaneous ICSRs with “Product” from worldwide source which included XXX ADRs, thereof XXX ADRs were serious. During the reporting interval the MAH received XXX spontaneous ICSRs with “Product” from worldwide source which included XXX ADRs, thereof XXX ADRs were serious. Table X: Reports of adverse drug reactions SOC PT e.g. Immune system disorders Urticaria No of serious events in interval No of nonserious events in interval No of serious events cumulatively No of nonserious events cumulatively Total number of ADRs Fatal cases: Please provide a short description of all fatal cases and provide company causality assessment for each case. 7. Summary of significant findings in the reporting period 7.1. Findings from completed clinical trials 7.2. Findings from ongoing clinical trials Product name 7.3. Findings from non-interventional studies 7.4. Findings from long-term follow-up 7.5. Findings from literature or independent studies 7.6. Findings from non-clinical studies 8. Non-clinical data 9. Literature The MAH should perform a search using his internal literature database. The report should include results of regular literature searches for defined active substances of the “product”. Please state who performs the literature searches, which literature databases are regularly searched (e.g. Medline, EMBASE) and discuss major publications. XXXX et al. Journal page year, short description of content XXXX et al. Journal page year, short description of content XXXX et al. Journal page year, short description of content 10. Lack of efficacy in controlled clinical trials Give a short overview of cases with treatment failure related to the IMP. 11. Late-Breaking Information Provide information on the safety or efficacy of “product” which was received after the data lock point of the report. 11.1. Risk minimisation measures/other actions 11.2. Ongoing risk minimisation measures 11.3. Newly initiated risk minimisation measures (reporting period) The following risk minimisation measures / actions were taken during the period covered by this safety report: Product name 11.4. Planned risk minimisation measures Please give a short overview on relevant measures for risk minimisation. E.g. ongoing / planned studies that can detect new risks, long term follow up measures ….. 12. Other Information e.g.: There has been no new information about abuse, misuse, or overdose of the product during the period covered by the PSUR. During the period of this report no case with medication error has been reported. 13. Signal evaluation and detection 13.1. Signal detection Please describe shortly the procedures in place for signal detection. 13.2. Closed signals Please add table(s) with signals that were closed during the reporting period. Give a short description of the signal and outline the reason for closing it. 13.3. Ongoing signals Please add table(s) with ongoing signals and describe nature of the signal. 13.4. New signals Please add table(s) with signals newly detected in the period and give a short description of the signal. 14. Risk evaluation 14.1. Summary of Safety Concerns Important Identified Risks Important Potential Risks Important identified interactions Missing Information 14.2. Characterisation and evaluation of risks Give a short description of all identified risks and evaluate risk associated cases. Give a short overview and state if the risk-benefit ratio has changed. Product name 14.3. Characterisation of important potential problems with the treatment and with certain populations Overview of current status Lack of effect Elderly patients Patients with renal and/or hepatic impairment Pregnant and lactating women Pediatric patients (<16yr/ <6yr/ <2yr) 15. Benefit/Risk Assessment 16. Overall Conclusions e.g.: The data provided in this PSUR describe sufficiently the safety profile of “product” in the interval and cumulatively. or The following new areas of concern related to the use of “product” in its licensed indications were identified: The benefit-risk evaluation remains positive because ……