pmic7244-sup-0001-figureS1
advertisement

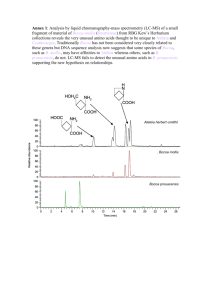
Supplemental Experimental Procedures Cell Culture Chang Liver cells were cultured at 37℃ in 5% CO2 in RPMI 1640 medium (Gibco, USA) supplemented with 10% newborn calf serum, 100 U/mL penicillin and 100μg/mL streptomycin (Gibco, USA). HepG2 cells, derived from human hepatocellular carcinoma, were cultured at 37℃ in 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, USA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100μg/mL streptomycin. Periodate Oxidation Chang Liver cells and HepG2 cells were all grown to ~90% confluence. The media were aspirated from each plate of cells and the cells were rinsed twice with 5 mL of PBS each time. Cells were oxidized with 3 mL 5 mM NaIO4 in oxidation buffer (100 mM NaAc, 150 mM NaCl, pH 5.5) in the dark at 25℃ for 1 h with gentle shaking, and then the periodate solution was removed by aspiration. The cells were harvested using a cell scraper (Corning, USA). Cells were pelleted and rinsed twice with 5 mL of oxidation buffer each time. Cell Lysis Cell pellets were homogenized in ice-cold 50 mM HEPES (pH 7.5), 65 mM dithiothreitol (DTT), 1 mM PMSF, 1% Triton and 2% cocktail (Sigma-Aldrich, USA) using an ultrasonic cell disrupter (Power was set at 400 W and time of ultrasonicaton was set at 3 s with 3 s intervals for 180 times). Homogenized lysates was centrifuged at 13000g at 4℃for 60 min and the supernatant was carefully collected. The protein concentration of the supernatant was determined by Bradford assay. N-linked Glycoprotein Enrichment by Hydrazide Chemistry The N-linked glycoproteins in each cell lysates were captured with hydrazide chemistry as described in a published protocol with minor modification[1]. 100 μL Bio-Rad Affi-Gel Hz hydrazide resin (bed volumn) was washed with coupling buffer (100 mM NaAc, 150 mM NaCl, pH 5.5) twice. About 1 mg of extracted proteins from both Chang liver cells and HepG2 cells were added to prewashed hydrazide resin. The coupling reaction was carried out overnight at room temperature with adequate shaking. The resin was washed with 1.5 M NaCl, methanol and 100 mM NH4HCO3 sequentially to remove non-specifically bound proteins. Release of Non-glycopeptides and N-glycopeptides After removal of NH4HCO3 solution from the last wash, the resin with captured glycoproteins was diluted with 8 M urea/100 mM NH4HCO3. The glycoproteins on the resin were reduced in 20 mM DTT at 37℃ for 2 h and carboxyamidomethylated in 40 mM iodoacetamide at room temperature for 40 min in the dark. The supernatant was removed and 100 mM triethyl ammonium bicarbonate (TEAB) (Sigma-Aldrich, USA) was added to the resin. Trypsin was added with a weight ratio of trypsin (Sigma-Aldrich, USA) to protein at 1/25 and incubated at 37℃overnight. The tryptic peptides were carefully collected by gentle centrifugation. The resin was further washed with 100 mM TEAB buffer. The supernatant was collected and combined with the tryptic peptides. The N-linked glycopeptides were released by adding 500 unit PNGase F (New England Biolabs, USA) in 100mM NH4HCO3 to the resin and incubating at 37℃overnight. The released N-glycopeptides were carefully collected by gentle centrifugation. Isotopic labeling of the Released Non-glycopeptides Non-glycopeptides from Chang Liver and HepG2 cells were labeled with light and heavy dimethyl respectively by an in-solution stable isotope dimethyl labeling approach according to the published protocol[2]. The separately labeled samples were mix together and desalted with a homemade C18 solid phase extraction column. After dried down in a Speed Vac (Thermo, San Jose, CA, USA), the sample was reconstituted in 0.1% FA aqueous solution for 2D nano LC-MS/MS analysis. Isotopic labeling of Both Non-glycopeptides and N-glycopeptides All the procedures before releasing N-glycopeptides were the same as the procedures described above. Then N-linked glycopeptides were released by adding 1μL of PNGase F (500 unit per μL) in 100 μL G7 buffer (50 mM sodium phosphate, pH=7.5) (New England Biolabs, USA) to the resin and incubating at 37℃ overnight. The released N-glycopeptides were carefully collected by gentle centrifugation. The resin was further washed with 100 μL G7 buffer. The supernatant was collected and combined with the N-glycopeptides. Both non-glycopeptides and N-glycopeptides from Chang Liver and HepG2 cells were labeled with light and heavy dimethyl respectively by an in-solution stable isotope dimethyl labeling approach according to the published protocol. The separately labeled samples were mix together and desalted with a homemade C18 solid phase extraction column. After dried down in a Speed Vac, the non-glycopeptides mixture was reconstituted in 0.1% FA aqueous solution for 2D nano LC-MS/MS analysis while the released N-glycopeptides mixture was for 1D nano LC-MS/MS analysis. Control Experiment: Differential Analysis of Cell Surface Glycoproteins between two HepG2 cells The control experiment was carried out to analyze the expression difference of cell surface glycoproteins between the two HepG2 cells. The procedures of control experiment are the same as the procedures described above, with enrichment for N-linked glycoprotein by hydrazide chemistry, glycopeptides for identification of glycoproteins and isotopically labeled non-glycopeptides for quantification of glycoproteins, except that the starting cell lines is the same cell line, i.e. HepG2 cells, not two different cell lines. N-linked Glycopeptide Enrichment by Hydrazide Chemistry About 1 mg of extracted proteins from both Chang Liver cells and HepG2 cells were first reduced in 20 mM DTT at 60℃ for 1 h and carboxyamidomethylated in 40 mM iodoacetamide at room temperature for 40 min in the dark. Then trypsin was added with a weight ratio of trypsin to protein at 1/25 and incubated at 37℃ overnight. The tryptic peptides were desalted with a homemade C18 solid phase extraction column. After dried down in a Speed Vac, the sample was reconstituted in 200ul coupling buffer (100 mM sodium acetate, 150 mM NaCl, pH 5.5) for glycopeptides enrichment. 50ul prewashed Bio-Rad Affi-Gel Hz hydrazide resin (bed volumn) was used for coupling reaction and the coupling reaction was carried out overnight at room temperature with adequate shaking. The resin was washed with 1 mL of 1.5 M NaCl, methanol and 100 mM NH4HCO3 sequentially three times to remove nonspecifically bound proteins. After removal of NH4HCO3 solution from the last wash, 1μL of PNGase F (500 unit per μL) in 200μL G7 buffer was added to the resin and incubated at 37℃ overnight. The supernatant was collected and labeled with light and heavy dimethyl respectively by an in-solution stable isotope dimethyl labeling approach according to the published protocol. The labeled deglycosylated peptides mixture was for 1D nano LC-MS/MS analysis. LC-MS/MS Analysis For each biological replicate (n=2), three technical replicates were analyzed by LC-MS/MS. The non-glycopeptides was analyzed by a 2D nano LC-MS/MS system as described previously[3] with minor change. The strong cation exchange (SCX) column was connected to a RP C18 column (12 cm × 75μm i.d) in tandem by a union. The SCX column was prepared by the same method as described by Wang et al.[4]. For the C18 capillary column, one end of the fused-silica capillary was manually pulled to a fine point of ~5μm with a flame torch, and then the C18 AQ beads (5μm, 12 nm) from Microm BioResources (Auburn, CA, USA) were packed until the packing section reached a length of 12 cm using a pneumatic pump. The RP C18 column was directly coupled with a LTQ-Orbitrap XL mass spectrometry (Thermo Finnigan, San Jose, CA, USA) with a nanospray source. The RPLC-MS/MS was performed on a nano-RPLC-MS/MS system. A Finnigan surveyor MS pump (Thermo Finnigan, San Jose, CA, USA) was used to deliver mobile phase. 0.1% Formic acid water solution (buffer A) and ACN with 0.1% formic acid (buffer B) were used for the generation of linear gradient for RPLC separation. 1000 mM NH4Ac (pH 2.7) as buffer C was used to generate step salt gradients to gradually elute the non-glycopeptides which were captured on SCX column onto the RP C18 analytical column after 25μL reconstituted non-glycopeptides were loaded automatically onto a phosphate SCX monolithic column (7 cm × 150μm i.d.). The serial stepwise salt concentrations of NH4Ac used for 10 step salt gradients were 50, 100, 150, 200, 250, 300, 350, 400, 500 and 1000 mM, respectively. Each salt step lasted 10 min and then the system was re-equilibrated with buffer A for another 15 min. After each salt elution, binary separation gradient from 5% to 35% in 100 min with a flow rate of ∼300 nL/min was started for reversed phase separation. The MS analysis was performed on LTQ-Orbitrap XL mass spectrometer at a resolution of 60 000. The temperature of the ion transfer capillary was set at 200℃. The spray voltage was set at 1.8 kV and normalized collision energy was set at 35%. One microscan was set for each MS and MS/MS scan. All MS and MS/MS spectra were acquired in the data-dependent mode. The mass spectrometer was set that one full MS scan was followed by six MS/MS scans on the six most intense ions by CID. The scan range was set from m/z 400 to m/z 2000. The target ion setting was 5e5, with a maximum fill-time of 500 ms. Minimum signal threshold set for MS/MS scan was 500. The dynamic exclusion function was set as follows: repeat count 2, repeat duration 30 s, and exclusion duration 60 s. The released N-glycopeptides were analyzed by 1D nano LC-MS/MS, in which the glycopeptides sample was loaded directly onto a RP C18 column and 120min binary separation gradient from 5% to 35% with a flow rate of ∼300 nL/min was used for separation. The MS analysis was performed as described above. Protein Identification and Quantification The process of the datasets of 2D nano LC-MS/MS was the same as described previously[3]. DTASupercharge (v2.0a7)[5] was applied to convert all the MS/MS spectra in one acquired raw file to a single *.mgf file. The *.mgf file was searched against a composite database including original and reversed human protein database of International Protein Index (ipi.human 3.17 fasta, including 60 234 entries, http://www.ebi.ac.uk/IPI/IPIhuman.html) by MASCOT Version 2.1 (Matrix Science, UK). Carboxamidomethylation (+57.0215 Da) of Cysteine residues was set as static modification and oxidation of methionine residues was searched as variable modification of +15.9949 Da. Also both light and heavy dimethylation of peptide N termini and the side chain of lysine residues were set as variable modification of +28.0313 Da and +32.0564 Da, respectively. Peptides were searched using fully tryptic cleavage constraints and a maximum of two missed cleavages sites were allowed for tryptic digestion. 10 ppm was set for the mass tolerances of parent masses and 0.8 Da for fragment masses. Peptides with Mascot score ≥25 (rank 1, P ≤0.05) were used for protein quantification. The false detection rate (FDR) was determined by equation of FDR=[2* FP/(FP+TP)]*100, where FP (false positive) is the number of peptides that were identified based on sequences in the reverse database component and TP (true positive) is the number of peptides that were identified based on sequences in the forward database component. FDR of each 2D nano LC-MS/MS analysis was controlled to be less than 1% by setting different Mascot score. A dimethyl-adapted version of MSQuant (v2.0a81) was applied for protein quantification. Peptide ratios were obtained by calculating the peak area of extracted ion chromatograms (XIC) of the light and heavy forms of the peptide using the monoisotopic peaks only and protein ratios were calculated from the average of the all quantified peptides[5, 6]. Then all the MSQuant outputs of all SCX fraction were imported into StatQuant (v1.2.2) and the quantified proteins were normalized against the log2 of the median ratio of all peptides quantified [6, 7]. Deamidation of both glutamines and asparagines (+0.9840 Da) were added as another variable modifications in the process of analyzing the results of 1D nano LC-MS/MS by MASCOT. The search result was exported in csv format and the Trans-Proteomics Pipeline (TPP, v4.3.1)[8] was used to convert raw file to ‘mzxml’ format. An in-house developed software Armone v1.0 beta 2[9] was applied to process the *.csv file together with *.mzxml file. FDR of the final identifications was controlled less than 1% by setting different filtering criterion. The proteins identified by the same peptides were grouped. To remove the redundancy of identifications, only one protein was kept for each group by using the following order as described by He et al.[10] : keep the proteins with annotation in Swiss-Prot and RefSeq_NP, keep the proteins with annotation in Swiss-Prot, and then keep the proteins with annotation in RefSeq_NP. If still over two proteins remained, the protein with longest sequence was kept as the unique entry in the group. Transmembrane domains were predicted by the common topology algorithm TMHMM v2.0, a hidden Markov model-based predictor for transmembrane helices in protein sequences[11]. Subcellular location analysis was performed using GoMiner[12] with the new database (version go_201202). Western Blotting Equal amount of extracted proteins from both Chang Liver cells and HepG2 cells were separated on a 12% SDS-PAGE gel and transferred to nitrocellulose membrane. Antibodies against basigin (EMMPRIN) and basal cell adhension molecule (BCAM) (Santa Cruz Biotechnology, CA, USA) were used to detect the protein of interest. Both primary antibodies were used with a dilution of 1:1000 and a second antibody against rabbit IgG (Santa Cruz Biotechnology, USA, 1:5000 dilution) was used for both proteins. Blots were developed with ECL western blotting reagent (Pierce, IL, USA). For western blot loading controls, β-actin monoclonal antibody (Cell Signaling Technology, USA) was used. Supplemental Figures Figure S1 (A) (B) (C) (D) (E) (F) Venn diagrams showing the overlapping and unique data of three runs of analyses. (A) 193 glycoproteins (RSD=2.1%, n=3) were identified in each run of three replicate runs of 1D LC-MS/MS in first biological replicate and more than 65% of total identified proteins in three runs can be identified in two or three runs. (B) 208 glycoproteins (RSD=5.5%, n=3) were identified in each run of three replicate runs of 1D LC-MS/MS in another biological replicate and more than 70% of total identified proteins in three runs can be identified in two or three runs. (C) 458 unique glycopeptides (RSD=2.4%, n=3) were identified in each run of three replicate runs of 1D LC-MS/MS in first biological replicate. (D) 508 unique glycopeptides (RSD=5.7%, n=3) were identified in each run of three replicate runs of 1D LC-MS/MS in another biological replicate. (E) 5379 unique tryptic peptides (RSD=10.4%, n=3) were identified in each run of three replicate runs of 2D LC-MS/MS in first biological replicate. (F) 5084 unique tryptic peptides (RSD=14.9%, n=3) were identified in each run of three replicate runs of 2D LC-MS/MS in another biological replicate. References [1] Zhang, H., Li, X. J., Martin, D. B., Aebersold, R., Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003, 21, 660-666. [2] Boersema, P. J., Raijmakers, R., Lemeer, S., Mohammed, S., Heck, A. J. R., Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4, 484-494. [3] Wang, F. J., Chen, R., Zhu, J., Sun, D. G., et al., A Fully Automated System with Online Sample Loading, Isotope Dimethyl Labeling and Multidimensional Separation for High-Throughput Quantitative Proteome Analysis. Anal. Chem. 2010, 82, 3007-3015. [4] Wang, F. J., Dong, J., Jiang, X. G., Ye, M. L., Zou, H. F., Capillary trap column with strong cation-exchange monolith for automated shotgun proteome analysis. Anal. Chem. 2007, 79, 6599-6606. [5] Mortensen, P., Gouw, J. W., Olsen, J. V., Ong, S. E., et al., MSQuant, an Open Source Platform for Mass Spectrometry-Based Quantitative Proteomics. J. Proteome Res. 2010, 9, 393-403. [6] Lemeer, S., Jopling, C., Gouw, J., Mohammed, S., et al., Comparative Phosphoproteomics of Zebrafish Fyn/Yes Morpholino Knockdown Embryos. Mol. Cell. Proteomics 2008, 7, 2176-2187. [7] van Breukelen, B., van den Toorn, H. W. P., Drugan, M. M., Heck, A. J. R., StatQuant: a post-quantification analysis toolbox for improving quantitative mass spectrometry. Bioinformatics 2009, 25, 1472-1473. [8] Pedrioli, P. G. A., Eng, J. K., Hubley, R., Vogelzang, M., et al., A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004, 22, 1459-1466. [9] Jiang, X. N., Ye, M. L., Cheng, K., Zou, H. F., ArMone: A Software Suite Specially Designed for Processing and Analysis of Phosphoproteome Data. J. Proteome Res. 2010, 9, 2743-2751. [10] He, P., He, H. Z., Dai, J., Wang, Y., et al., The human plasma proteome: Analysis of Chinese serum using shotgun strategy. Proteomics 2005, 5, 3442-3453. [11] Krogh, A., Larsson, B., von Heijne, G., Sonnhammer, E. L. L., Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 2001, 305, 567-580. [12] Zeeberg, B. R., Feng, W. M., Wang, G., Wang, M. D., et al., GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003, 4.